The first law of thermodynamics considers interpretations of the law of conservation of energy and how it applies to the system (the reaction under consideration). The second law, however, is interested in the measurement of randomness in a system, the entropy. Before we get down to the nitty-gritty on what that actually means, let's examine some ways in which entropy influences the world around us.
Example 1: Cup of Tea
Say you add an ice cube to your cup of tea. Aside from the wonderful crackling noise the ice cube makes as it melting, what is really going on? First of the enthalpy of the reaction is increasing as the ice cube requires heat to melt. And then, the entropy is also increasing.
Why? Well, in a solid the molecules have less degrees of rotation, as they solely vibrate in a dense structure, but when they melt to become liquid water, the molecules can move around and slide past one another, which is why if you tip over your tea it will spill all over the floor and will make the mess that it does. These further degrees of freedom increase the amount of randomness in the system. And hence, liquids have greater entropy than solids.
Now, let's consider the steam coming off of your tea cup. As you may have anticipated, gases of a substance have even higher entropy than liquids, because the molecules have greater degrees of freedom than liquids.
So in summary:
$$entropy_{solid} < entropy_{liquid} < < entropy_{gas}$$
More degrees of freedom = more entropy
Example 2: Denaturing DNA
Denaturing DNA = breaking DNA from its classic double helix shape into two separate strands.
Often in the laboratory denaturation of DNA is done in a test tube by increasing the temperature so that the hydrogen bonds that connect the bases (A,T,G,C) break. This is an endothermic process as it requires input of energy, high temperature and results in something with more entropy (2 strands instead of 1).
Denaturing DNA is also a reversible process, so if you decrease the temperature, the strands will re-anneal to the double-stranded helix structure. This can be investigated more clearly by checking out the Gibb's Free Energy equation $G=H-TS$, which I will discuss in a later post.
Basically, the take home message is that for any reaction, there is a dialogue between the favourable enthalpy and favourable entropy conditions, which will determine whether the reaction is spontaneous or not.
In example 1 with the tea cup, the ice melting and the steam rising, was all spontaneous at room temperature. However, in example 2, this is not as clear. The reaction is temperature dependent, as temperature is the factor that mediates the relationship between the enthalpy and entropy of the reaction.
But now you are probably wondering: what are the favourable enthalpy and favourable entropy conditions for a reaction to occur spontaneously? Fair enough.
For a reaction to occur spontaneously, the it prefers to have negative enthalpy, which is an exothermic process (releases heat into the environment).
"Okay," you say, "But what if I have a giant stick of dynamite in my hands? That releases a lot of heat because it explodes. So why isn't this reaction spontaneous? Clearly it is an exothermic reaction."
Good point. Recall a potential energy diagram for an exothermic process. Some reactions are required to overcome their activation energy for the reactants to turn into the products. Here is a quick 30 second video to trigger your memory if you have forgotten this concept. Activation Energy Definition So in the case of dynamite, it will not spontaneously combust because it requires a certain input of energy - the spark to ignite it - to blow up and overcome its activation energy.
So, we've covered the spontaneous conditions for enthalpy, but what about entropy? The system is favours states of greater disorder, so the entropy increases positively. A way representing this formally is defining our system (because we can take a system to be as small or as big as we like, as we are the ones who define it) as the universe. Since the universe is an isolated system, we know that
$$\Delta{S}_{universe}\geq{0}$$
Now, this explains why certain reactions with respect to a smaller system can occur at all. As long as the $\Delta{S}_{universe}$ remains positive, which it will because of the massive size of the system, a small negative blip where $\Delta{S}_{system}\leq{0}$, will simply not matter as it is insignificant in the grand scheme of things. We represent this by
$$\Delta{S}_{universe}+\Delta{S}_{system}\geq{0}$$
or
$$\Delta{S}_{system}+\Delta{S}_{surroundings}\geq{0}$$
Also, note that a reversible process is defined by
$$\Delta{S}_{system}=-\Delta{S}_{surroundings}$$
All right, this leads us to example 3.
Example 3: An Endothermic Reaction
$$\text{N}_2\text{O}_4\longrightarrow 2\text{NO}_2$$
So what is happening here? Dinitrogen dioxide on the left hand side of the equation is reacting to become the product nitrogen monoxide. The key thing to notice is that there is only 1 mole of reactant to 2 moles of products, so even though this reaction absorbs energy and is endothermic, and hence should be unfavourable, the positive increase in entropy (more products than reactants, therefore more disorder) favours the reaction to proceed. In this case, the drive of the entropy overcomes the enthalpy of the reaction.
In the next post I will discuss a statistical representation of entropy, so stay tuned! :)
Tuesday, 20 November 2012
Monday, 19 November 2012
An Introduction to Entropy
What is entropy? A measure of disorder can seem abstract and somewhat difficult to envision at times. Can I really blame the state of my apartment with unwashed dishes and dirty laundry strewn on the floor on its agency - or does this have to do more with the subjective nature of personality traits? Can one really measure quantitively disorder when colloquially we refer to it in quantitative terms?
I am very fond of the topic of entropy. In high school, I used to tell horror stories about the eventual heat death of the universe ad naseum. Eventually all of the particles would exist in a solid, perfect crystal state, where no energy could rescue them from their eternal plight.
My musician father was fascinated by entropy while listening to CBC radio program Ideas. Many a skype conversation, he will pounce the topic upon me, and question continuously its definition and properties. Once we devised a system of cells in the human body, existing under ideal conditions (eg: no negative effects of the external environment such as a blow to some tissue, or disease), with only the natural aging process as a factor. Applying the statistical explanation of entropy, we were able to determine it was statically unlikely for all of the cells in an common region of the body to spontaneously die, when otherwise undisturbed.
Walking one evening with a someone special, in the crisp fall Montréal air, we discussed how if you have an empty chamber attached to a flask filled with particles of gas, and opened the flask to the empty chamber, the gas particles would distribute themselves so they filled every state in the chamber. The movement of our postulated system of order to one of disorder mirrored that of the Big Bang. We found it disturbing that the transition from order to disorder was mediated by an external event of the person opening the stopcock. Does this part of the analogy translate over to the scenario of the Big Bang as well or not? Where did the initial stimulus for the start of the universe come from?
Entropy can lead to interesting conversations with people of all different backgrounds, and also contribute to an enhanced understanding of the behaviour of the universe. For these reasons, I am going to develop the concept of entropy over a series of posts, discussing both statistical and thermodynamic interpretations of entropy, and employing both qualitative and quantitative means throughout my explanations.
These posts will be dedicated to my dad because I have always promised to explain entropy throughly to him, but we never have sat down properly to discuss it.
I am very fond of the topic of entropy. In high school, I used to tell horror stories about the eventual heat death of the universe ad naseum. Eventually all of the particles would exist in a solid, perfect crystal state, where no energy could rescue them from their eternal plight.
My musician father was fascinated by entropy while listening to CBC radio program Ideas. Many a skype conversation, he will pounce the topic upon me, and question continuously its definition and properties. Once we devised a system of cells in the human body, existing under ideal conditions (eg: no negative effects of the external environment such as a blow to some tissue, or disease), with only the natural aging process as a factor. Applying the statistical explanation of entropy, we were able to determine it was statically unlikely for all of the cells in an common region of the body to spontaneously die, when otherwise undisturbed.
Walking one evening with a someone special, in the crisp fall Montréal air, we discussed how if you have an empty chamber attached to a flask filled with particles of gas, and opened the flask to the empty chamber, the gas particles would distribute themselves so they filled every state in the chamber. The movement of our postulated system of order to one of disorder mirrored that of the Big Bang. We found it disturbing that the transition from order to disorder was mediated by an external event of the person opening the stopcock. Does this part of the analogy translate over to the scenario of the Big Bang as well or not? Where did the initial stimulus for the start of the universe come from?
Entropy can lead to interesting conversations with people of all different backgrounds, and also contribute to an enhanced understanding of the behaviour of the universe. For these reasons, I am going to develop the concept of entropy over a series of posts, discussing both statistical and thermodynamic interpretations of entropy, and employing both qualitative and quantitative means throughout my explanations.
These posts will be dedicated to my dad because I have always promised to explain entropy throughly to him, but we never have sat down properly to discuss it.
Sunday, 18 November 2012
Ideal Gases: Reversible Adiabatic Process
For a reversible adiabatic process, the system is moving between two different isotherms, so the temperature is changing. Also, the both volume and pressure are changing. So $P_1V_1T_1 \rightarrow P_2V_2T_2$.
The work done in an adiabatic process is less than that of an isothermal process, as an isothermal process is the maximum work possible to be done on the system. $w_{adiabatic} < w_{isothermal}$
For a reversible adiabatic process, there is no heat transfer to the system by definition, thus $q=0$. Since $\Delta{U}=q+w$, then $\Delta{U}=w=-PdV$.
For the following derivation, to make our lives a bit easier, we are going to consider the case where $n=1$. $$dU=-PdV$$ $$C_vdT=-PdV$$ $$C_vdT+PdV=0$$ $$C_vdT+RTdV/V=0$$ $$\text{as } P=RT/V$$ $$\text{divide equation by } T$$ $$C_vdT/T+RT/TdV/V=0/T$$ $$C_vdT/T+RdV/V=0$$ $$C_v\int_{T_1}^{T_2}dT/T+R\int_{V_1}^{V_2}dV/V=0$$ $$C_vln(T_2/T_1)+Rln(V_2/V_1)=0$$ $$\text{recall that }R=C_p-C_v$$ $$\text{divide by }C_v$$ $$C_v/C_vln(T_2/T_1)+(C_p-C_v)/C_vln(V_2/V_1)=0$$ $$ln(T_2/T_1)+(C_p/C_v+1)ln(V_2/V_1)=0$$ $$\text{let } C_p/C_v =\gamma$$ $$ln(T_2/T_1)+ln(V_2/V_1)^{\gamma-1}=0$$ $$ln(T_2/T_1)=ln(V_1/V_2)^{\gamma-1}$$ $$T_2/T_1=(V_1/V_2)^{\gamma-1}$$ $$\text{since } PV=RT$$ $$(P_2V_2)/(P_1V_1)=(V_1/V_2)^{\gamma-1}$$ $$P_2/P_1=(V_1/V_2)^{\gamma-1}(V_1/V_2)$$ $$P_2/P_1=(V_1/V_2)^{\gamma}$$ $$P_1V_1^{\gamma}=P_2V_2^{\gamma} \text{ where } \gamma = C_p/C_v$$
To check our work, we can see if this applies to the ideal gas law. $$P_1V_1^{\gamma}=P_2V_2^{\gamma}$$ $$PV^{\gamma}=RTV^{\gamma}/V$$ $$PV^{\gamma}=RTV^{\gamma-1}$$ $$PV^{\gamma}/V^{\gamma}=RTV^{\gamma-1}/V^{\gamma}$$ $$P=RT/V$$ The gamma term can also be related to the number of degrees of freedom,$f$, in a gas. $$\gamma = C_p/C_v = \Delta{H}/\Delta{U}=(f+2)R/fR= (f+2)/f$$
To check out $\Delta{H}$, the entropy, we consider $$\Delta{H}=\Delta{U}+\Delta{PV}=w+q+\Delta{PV}=w+R\Delta{T}$$ $$\Delta{H}=C_v\Delta{T}+R\Delta{T}$$ $$\Delta{H}=(C_v+R)\Delta{T}$$ $$\Delta{H}=C_p\Delta{T}$$
In summary:
$$q=0$$ $$\Delta{U}=w=-PdV$$ $$\Delta{H}=C_p\Delta{T}$$ $$P_1V_1^{\gamma}=P_2V_2^{\gamma} \text{ where } \gamma = C_p/C_v$$
The work done in an adiabatic process is less than that of an isothermal process, as an isothermal process is the maximum work possible to be done on the system. $w_{adiabatic} < w_{isothermal}$
For a reversible adiabatic process, there is no heat transfer to the system by definition, thus $q=0$. Since $\Delta{U}=q+w$, then $\Delta{U}=w=-PdV$.
For the following derivation, to make our lives a bit easier, we are going to consider the case where $n=1$. $$dU=-PdV$$ $$C_vdT=-PdV$$ $$C_vdT+PdV=0$$ $$C_vdT+RTdV/V=0$$ $$\text{as } P=RT/V$$ $$\text{divide equation by } T$$ $$C_vdT/T+RT/TdV/V=0/T$$ $$C_vdT/T+RdV/V=0$$ $$C_v\int_{T_1}^{T_2}dT/T+R\int_{V_1}^{V_2}dV/V=0$$ $$C_vln(T_2/T_1)+Rln(V_2/V_1)=0$$ $$\text{recall that }R=C_p-C_v$$ $$\text{divide by }C_v$$ $$C_v/C_vln(T_2/T_1)+(C_p-C_v)/C_vln(V_2/V_1)=0$$ $$ln(T_2/T_1)+(C_p/C_v+1)ln(V_2/V_1)=0$$ $$\text{let } C_p/C_v =\gamma$$ $$ln(T_2/T_1)+ln(V_2/V_1)^{\gamma-1}=0$$ $$ln(T_2/T_1)=ln(V_1/V_2)^{\gamma-1}$$ $$T_2/T_1=(V_1/V_2)^{\gamma-1}$$ $$\text{since } PV=RT$$ $$(P_2V_2)/(P_1V_1)=(V_1/V_2)^{\gamma-1}$$ $$P_2/P_1=(V_1/V_2)^{\gamma-1}(V_1/V_2)$$ $$P_2/P_1=(V_1/V_2)^{\gamma}$$ $$P_1V_1^{\gamma}=P_2V_2^{\gamma} \text{ where } \gamma = C_p/C_v$$
To check our work, we can see if this applies to the ideal gas law. $$P_1V_1^{\gamma}=P_2V_2^{\gamma}$$ $$PV^{\gamma}=RTV^{\gamma}/V$$ $$PV^{\gamma}=RTV^{\gamma-1}$$ $$PV^{\gamma}/V^{\gamma}=RTV^{\gamma-1}/V^{\gamma}$$ $$P=RT/V$$ The gamma term can also be related to the number of degrees of freedom,$f$, in a gas. $$\gamma = C_p/C_v = \Delta{H}/\Delta{U}=(f+2)R/fR= (f+2)/f$$
To check out $\Delta{H}$, the entropy, we consider $$\Delta{H}=\Delta{U}+\Delta{PV}=w+q+\Delta{PV}=w+R\Delta{T}$$ $$\Delta{H}=C_v\Delta{T}+R\Delta{T}$$ $$\Delta{H}=(C_v+R)\Delta{T}$$ $$\Delta{H}=C_p\Delta{T}$$
In summary:
$$q=0$$ $$\Delta{U}=w=-PdV$$ $$\Delta{H}=C_p\Delta{T}$$ $$P_1V_1^{\gamma}=P_2V_2^{\gamma} \text{ where } \gamma = C_p/C_v$$
Ideal Gases: Reversible Isothermal Process
Consider system with an ideal frictionless piston to apply and reduce pressure and change the volume. Recall that for an isothermal process that is reversible, the $P_{gas}=P_{ext}$. In this case the work in = heat out. Also, there is no net change in both $\Delta{U}$ and $\Delta{H}$, because the process occurs along the same isotherm, so $\Delta{T}=0$, as $T_1=T_2$.
Therefore (a bit redundant, but we'll show the equations anyway):
$$\Delta{U}=C_v(T_2-T_1)=0$$ $$\Delta{H}=C_p(T_2-T_1)=0$$
Recall, the equation of work of an ideal gas $$w=RTln(V_1/V_2)$$
Since the heat out is equal to the work in, the heat is given by $$q=-w=-RTln(V_1/V_2)=RTln(V_2/V_1)$$
Note about the Concentration:
When the number of moles are not equal to one, we cannot ignore the $n$ term. So when have some $P_1=n_1/V_1RT=C_1RT$ and $P_2=n_2/V_2RT=C_2RT$ where $C$ is the concentration.
Therefore, the ideal gas equation for work can also be expressed by $$w=nRTln(V_1/V_2)=NRTln(P_2/P_1)\text{ as } P_2/P_1=V_1/V_2$$ and $$w=nRTln(C_2/C_1)$$
These equations come directly from the definitions of the terms. If they seem a bit confusing, refer to earlier physical chemistry posts on this blog, which can be found via the table of contents. Or if not, check wikipedia or something. :)
Therefore (a bit redundant, but we'll show the equations anyway):
$$\Delta{U}=C_v(T_2-T_1)=0$$ $$\Delta{H}=C_p(T_2-T_1)=0$$
Recall, the equation of work of an ideal gas $$w=RTln(V_1/V_2)$$
Since the heat out is equal to the work in, the heat is given by $$q=-w=-RTln(V_1/V_2)=RTln(V_2/V_1)$$
Note about the Concentration:
When the number of moles are not equal to one, we cannot ignore the $n$ term. So when have some $P_1=n_1/V_1RT=C_1RT$ and $P_2=n_2/V_2RT=C_2RT$ where $C$ is the concentration.
Therefore, the ideal gas equation for work can also be expressed by $$w=nRTln(V_1/V_2)=NRTln(P_2/P_1)\text{ as } P_2/P_1=V_1/V_2$$ and $$w=nRTln(C_2/C_1)$$
These equations come directly from the definitions of the terms. If they seem a bit confusing, refer to earlier physical chemistry posts on this blog, which can be found via the table of contents. Or if not, check wikipedia or something. :)
Ideal Gases: Isochoric Process
An isochoric process occurs where the volume is constant, $V_1=V_2$. This means that for the ideal gas law, $PV=nRT$, $T \propto P$.
Since $\Delta{V}=0$, then the relationship $w=-P\Delta{V}=0$ as well.
Examining the heat at constant volume, we find
$q_v=C_v\int_{T_1}^{T_2}dT=C_v(T_2-T_1)$
Now, when we look at the internal energy
$\Delta{U}=q_v+w=q_v=C_v(T_2-T_1)$
And finally, the entropy for when $n=1$, with
$$\Delta{H}=\Delta{U}+\Delta(PV)$$ $$\Delta{H}=q_v+\Delta{RT}$$ $$\Delta{H}=C_v(T_2-T_1)+R(T_2-T_1)$$ $$\Delta{H}=(C_v+R)(T_2-T_1)$$
So, in summary, for an isochoric process $$w_{rev}=0$$ $$q_v=\delta{U}=C_v(T_2-T_1)$$ $$\Delta{H}=(C_v+R)(T_2-T_1)$$
Note, that like for isobaric processes, isochoric processes occur on two different isotherms, as the temperature is not constant. These two isotherms of the isochoric process can be the same isotherms as for an isobaric process, however the work and heat supplied vary.
Since $\Delta{V}=0$, then the relationship $w=-P\Delta{V}=0$ as well.
Examining the heat at constant volume, we find
$q_v=C_v\int_{T_1}^{T_2}dT=C_v(T_2-T_1)$
Now, when we look at the internal energy
$\Delta{U}=q_v+w=q_v=C_v(T_2-T_1)$
And finally, the entropy for when $n=1$, with
$$\Delta{H}=\Delta{U}+\Delta(PV)$$ $$\Delta{H}=q_v+\Delta{RT}$$ $$\Delta{H}=C_v(T_2-T_1)+R(T_2-T_1)$$ $$\Delta{H}=(C_v+R)(T_2-T_1)$$
So, in summary, for an isochoric process $$w_{rev}=0$$ $$q_v=\delta{U}=C_v(T_2-T_1)$$ $$\Delta{H}=(C_v+R)(T_2-T_1)$$
Note, that like for isobaric processes, isochoric processes occur on two different isotherms, as the temperature is not constant. These two isotherms of the isochoric process can be the same isotherms as for an isobaric process, however the work and heat supplied vary.
Saturday, 17 November 2012
Ideal Gases: Isobaric Process
Ideal Gases Isobaric Processes
When examining our favourite ideal gas law of $PV=nRT$, we are going to consider a case where the pressure remains constant. Such a case is referred to as an isobaric process, and $P_1 = P_2$.
For example, you have a balloon, with some initial pressure, volume, and temperature. Say that you leave the balloon outside in the cold Montréal winter overnight. In the morning, you would probably see that the balloon shrank. What happened? Well, the atmospheric pressure remained constant, but the temperature changed, dropping to some infernal degree in the negatives. So since the pressure and moles can be eliminated from the ideal gas equation, we have the relationship $V\propto T$. As the temperature decreased overnight, so must the volume to compensate.
Now, enough of that. Let's investigate further to see what happens to the PV work in an isobaric process:
$$w_{rev} = -\int_{V_1}^{V_2}P_1dV
$$ $$w_{rev}= -P_1(V_2-V_1)=+P_1(V_1-V_2)
$$ $$w_{rev}=P_1(RT_1/P_1-RT_2/P_1)
$$ $$w_{rev}=R(T_1-T_2)
$$
Note that $V_1>V_2$, and also $n=1$.
The heat is given by $heat=q_p =\int_{T_1}^{T_2}C_Pd = C_p(T_2-T_1)$
The change in internal energy is given by
$$\Delta{U}=q_p+w$$ $$\Delta{U}=C_p(T_2-T_1)-R(T_2-T_1)$$ $$\Delta{U}=(C_p-R)(T_2-T_1)$$ $$\Delta{U}=C_v(T_2-T_1)$$
In summary for an isobaric process, $$q_p=C_p(T_2-T_1)$$ $$w_{rev}=-R(T_2-T_1)$$ $$\Delta{H} = q_p = C_p(T_2-T_1)$$ $$\Delta{U}=C_v(T_2-T_1)$$
This process occurs on two different isotherms.
When examining our favourite ideal gas law of $PV=nRT$, we are going to consider a case where the pressure remains constant. Such a case is referred to as an isobaric process, and $P_1 = P_2$.
For example, you have a balloon, with some initial pressure, volume, and temperature. Say that you leave the balloon outside in the cold Montréal winter overnight. In the morning, you would probably see that the balloon shrank. What happened? Well, the atmospheric pressure remained constant, but the temperature changed, dropping to some infernal degree in the negatives. So since the pressure and moles can be eliminated from the ideal gas equation, we have the relationship $V\propto T$. As the temperature decreased overnight, so must the volume to compensate.
Now, enough of that. Let's investigate further to see what happens to the PV work in an isobaric process:
$$w_{rev} = -\int_{V_1}^{V_2}P_1dV
$$ $$w_{rev}= -P_1(V_2-V_1)=+P_1(V_1-V_2)
$$ $$w_{rev}=P_1(RT_1/P_1-RT_2/P_1)
$$ $$w_{rev}=R(T_1-T_2)
$$
Note that $V_1>V_2$, and also $n=1$.
The heat is given by $heat=q_p =\int_{T_1}^{T_2}C_Pd = C_p(T_2-T_1)$
The change in internal energy is given by
$$\Delta{U}=q_p+w$$ $$\Delta{U}=C_p(T_2-T_1)-R(T_2-T_1)$$ $$\Delta{U}=(C_p-R)(T_2-T_1)$$ $$\Delta{U}=C_v(T_2-T_1)$$
In summary for an isobaric process, $$q_p=C_p(T_2-T_1)$$ $$w_{rev}=-R(T_2-T_1)$$ $$\Delta{H} = q_p = C_p(T_2-T_1)$$ $$\Delta{U}=C_v(T_2-T_1)$$
This process occurs on two different isotherms.
Wednesday, 31 October 2012
Look the equations on this Blog for Entropy will be AMAZING! :D
Consider $\sigma : \mathbb{R}\to\mathbb{R}$ a linear transformation.
$$
\int_0^\infty e^{-x}dx = 1
$$
S=dh/dT
$S=\frac{dH}{dT}$
$\sum_{i=1}^\infty \frac{1}{e^i}$
Thursday, 25 October 2012
Enhancer Odds and Sods
Transfection Assay: Activity of Enhancers
Comparison of a virus with a specific gene and an enhancer, and a virus with a specific gene and no enhancer can reveal the activity of the enhancer by seeing qualitatively how much more of the mRNA is transcribed by the trial with the enhancer present.
Eukaryotic Genes Are Regulated By Many Transcriptional Control Elements
Pretty much the what the heading of this sentence says. Some of elements include the TATA box, the promoter proximal elements, enhancers - and remember enhancers can be anywhere!
Something About Yeast
A common regulatory element that acts like an enhancer in yeast is called UAS. The TATA box in yeast is 90 base pairs from start site. Your life is now complete from knowing these details. :)
Comparison of a virus with a specific gene and an enhancer, and a virus with a specific gene and no enhancer can reveal the activity of the enhancer by seeing qualitatively how much more of the mRNA is transcribed by the trial with the enhancer present.
Eukaryotic Genes Are Regulated By Many Transcriptional Control Elements
Pretty much the what the heading of this sentence says. Some of elements include the TATA box, the promoter proximal elements, enhancers - and remember enhancers can be anywhere!
Something About Yeast
A common regulatory element that acts like an enhancer in yeast is called UAS. The TATA box in yeast is 90 base pairs from start site. Your life is now complete from knowing these details. :)
Deletion Analysis: Mapping Enhancers
Deletion Analysis works to find enhancers that can be many kilobases away from the start site of a gene. By taking a longer stretch of DNA sequence, and using a restriction enzyme to cut at various sites of the sequence, when the modified sequence inserted into a plasmid vector and placed within the cell, the amount of reporter gene indicates whether transcription has been affected. Comparison between transcription of various modified lengths of the sequence from longest sequence to shortest sequence reveals the location of the enhancer in an interval of bases. This technique is not as precise, but it still finds the region of the enhancer element, much in the same manner that a linker mutation technique does, by seeing where transcription stops. The region of the enhancer element is found within the difference of the bases of the closest longer sequence trial and the sequence where the transcription stops.
Linker Scanning Mutations
To locate promoter proximal elements in a short stretch of DNA up to around 100 base pair upstream of the TATA box, an experiment involving linker-scanning mutations can be used. A region of DNA that sequences a reporter gene is cloned several times. The clones are altered by addition of scrambled nucleotide sequences known as linker scanning mutations that are introduced from one end of the region of the sequence to the other. This is only done in a short stretch of the DNA to pinpoint the exact region of the promoter proximal elements. Then when the sequence is placed back into the cell via a plasmid vector, the rate of transcription (how much mRNA is produced) is measured by the amount of proliferation of the reporter gene. If transcription occurs, the region does not contain a promoter proximal element. If transcription has stopped or is reduced, the region of the linker mutation is that of a promoter proximal element, and the bases can then be identified.
Eukaryotic Gene Expression: The TATA Box and Other Promoters and Enhancers
Meet the TATA Box
First thing is that structure-wise this structure doesn't have too many silly nomenclature tricks up its sleeve. For once, the name is what it says it is, a TA rich region on a DNA sequence. Functionally, it positions the RNA polymerase on the DNA sequence for transcription, acting similarly to an E coli promoter. The TATA box is located around -35 to -25 base pairs upstream of the start site. So if some clever multiple choice question asks "Is the start site at the TATA box?" the answer is "No, it is downstream." "Do all genes have a TATA Box?" Again, "No, only those that have high rates of transcription in the cell." We will see plenty of other wonderful ways genes can be transcribed without a TATA box, albeit not in much detail. They will be discussed shortly.
How TATA is your TATA Box?
The TATA box is called a consensus sequence, and it is highly conserved among various genes in various organisms. However, each of the bases (A, T, and also G, and C) have a certain frequency of being in the ideal location "TATATATA" of the TATA box.
The third base has a 100% frequency of having a T, but the other bases are not as clear cut, with the first base being 83% likely of having a T, the second being 91% likely of having an A, and the others having 100%, 95%, 33%, 97%, 36% and 41% for the ideal base be it T or A respectively. There is a 40% probability of having a G in the last position.
This looks like memorization hell, and quite frankly it is, but the take-home message (which never shows up in multiple choice for this course for the record, oops that might have been sass) says the following: the TATA box is highly conserved, however different bases may be present in its sequence. I take back what I first said about the nomenclature, a better name would be the "most likely but not quite always TATA box."
Alternatives to the TATA Box
Initiator Element
Not much is known or considered important to relay at the undergraduate level about initiator elements except that a C is found in the -1 position and an A is found in the +1 position. I am really not sure how they did experimental testing to figure out these details, except it really must occur with a significant (or relatively significant) degree of frequency in the sequence, so if anyone knows and the explanation isn't too complicated that would be cool.
CpG Islands
These are CG rich areas of 20-50 base pairs within 100 base pairs of the start site region of a gene. These genes often have multiple start sites for transcription in a 20-200 bp region, and have neither a TATA box, nor initiator elements.
Promoter Proximal Elements
These are sequences within 100-200 base pairs of the start sequence that aren't the TATA box or any of the above sequences mentioned. They can be cell-type specific (not universally conserved).
Enhancers
Enhancers can be quite far away from the gene they enhance - even greater than 50 kilobases away! Their location may be upstream from the promoter, downstream from the promoter, within an intron, or downstream of the final exon of the gene. As one of my favourite Beatles songs likes to say, they can be quite literally be "here, there, and everywhere." The direction of the enhancer doesn't matter! Also, like promoter Proximal Elements, they are often cell type specific.
Difference between Promoter Proximal Elements and Enhancers?
Recall that it is with a human categorical bias that we organize these components of the cell. So, the distinction between promoter proximal elements and enhancers is not clear cut. For the purposes of the course I am taking, if something is within 100-200 base pair of the start sequence and helps initiate transcription, we'd probably call it a promoter proximal element - but who knows it could just as easily be an enhancer, or perhaps it is both!
**The next post will continue with an explanation about finding Promoter Proximal Elements with linker scanning mutations, and deletion analysis, and then there will also be another post about Enhancers and their effects on transcription.
First thing is that structure-wise this structure doesn't have too many silly nomenclature tricks up its sleeve. For once, the name is what it says it is, a TA rich region on a DNA sequence. Functionally, it positions the RNA polymerase on the DNA sequence for transcription, acting similarly to an E coli promoter. The TATA box is located around -35 to -25 base pairs upstream of the start site. So if some clever multiple choice question asks "Is the start site at the TATA box?" the answer is "No, it is downstream." "Do all genes have a TATA Box?" Again, "No, only those that have high rates of transcription in the cell." We will see plenty of other wonderful ways genes can be transcribed without a TATA box, albeit not in much detail. They will be discussed shortly.
How TATA is your TATA Box?
The TATA box is called a consensus sequence, and it is highly conserved among various genes in various organisms. However, each of the bases (A, T, and also G, and C) have a certain frequency of being in the ideal location "TATATATA" of the TATA box.
The third base has a 100% frequency of having a T, but the other bases are not as clear cut, with the first base being 83% likely of having a T, the second being 91% likely of having an A, and the others having 100%, 95%, 33%, 97%, 36% and 41% for the ideal base be it T or A respectively. There is a 40% probability of having a G in the last position.
This looks like memorization hell, and quite frankly it is, but the take-home message (which never shows up in multiple choice for this course for the record, oops that might have been sass) says the following: the TATA box is highly conserved, however different bases may be present in its sequence. I take back what I first said about the nomenclature, a better name would be the "most likely but not quite always TATA box."
Alternatives to the TATA Box
Initiator Element
Not much is known or considered important to relay at the undergraduate level about initiator elements except that a C is found in the -1 position and an A is found in the +1 position. I am really not sure how they did experimental testing to figure out these details, except it really must occur with a significant (or relatively significant) degree of frequency in the sequence, so if anyone knows and the explanation isn't too complicated that would be cool.
CpG Islands
These are CG rich areas of 20-50 base pairs within 100 base pairs of the start site region of a gene. These genes often have multiple start sites for transcription in a 20-200 bp region, and have neither a TATA box, nor initiator elements.
***********************
Promoter Proximal Elements
These are sequences within 100-200 base pairs of the start sequence that aren't the TATA box or any of the above sequences mentioned. They can be cell-type specific (not universally conserved).
Enhancers
Enhancers can be quite far away from the gene they enhance - even greater than 50 kilobases away! Their location may be upstream from the promoter, downstream from the promoter, within an intron, or downstream of the final exon of the gene. As one of my favourite Beatles songs likes to say, they can be quite literally be "here, there, and everywhere." The direction of the enhancer doesn't matter! Also, like promoter Proximal Elements, they are often cell type specific.
Difference between Promoter Proximal Elements and Enhancers?
Recall that it is with a human categorical bias that we organize these components of the cell. So, the distinction between promoter proximal elements and enhancers is not clear cut. For the purposes of the course I am taking, if something is within 100-200 base pair of the start sequence and helps initiate transcription, we'd probably call it a promoter proximal element - but who knows it could just as easily be an enhancer, or perhaps it is both!
**The next post will continue with an explanation about finding Promoter Proximal Elements with linker scanning mutations, and deletion analysis, and then there will also be another post about Enhancers and their effects on transcription.
Monday, 22 October 2012
The Epic Tale of the Man who Stepped on Glass
This post took awhile to write and is hopefully accurate. After sifting through lecture notes, my textbook, and internet articles, "The Epic Tale of the Man who Stepped on Glass" compiles the entire immune response as required for understanding of someone enrolled in a first-ever physiology class. While reading, I found that often lists of cells would be provided and then lists of processes. This "tale" takes on both concurrently! Please do note that the focus is on bacterial infection, and not viral infection. However, there is a note at the end, which explains a bit about viral infection as well.
Once upon a time, there was a man. He may have been a good man or a bad man, I'm not entirely sure, however, the qualifier in this case is irrevelant. On one fateful day the man accidentally (or consciously, but it is highly unlikely this was the case) stepped on a piece of broken glass. The glass had shattered onto the ground, and as he was barefoot, and one of the fragments pierced his skin. http://www.youtube.com/watch?v=Rw0eIG_9tLY
Apart from the man's howl of anguish as the blood dripped down the arch of his foot, other courses of action were taking place in the man's body. Yes, his foot was throbbing and swollen, but at the cellular level, things were at work. The glass had broken through the first line of defence, the coverings of the body. The skin and the mucous membranes usually provide an unpleasant environment for the living conditions of microorganisms, but the glass had successfully penetrated the skin. Little did the man know that the glass had been infected by a bacteria, so the next line of defence had to be called into action.
The fixed tissue macrophage, present at the site of the infection, is the first cell to be involved in the innate immune response. They have toll-like receptors (TLRs), which are attracted by the PAMPs produced by the bacteria. PAMPs, pathogen-associated molecular patterns, are lipopolysaccrides or peptidylglycan molecules present on the surface of bacteria, which are absent in mammals, and are therefore considered foreign. The fixed tissue macrophage will take up some bacteria by phagocytosis, and emit a signal called the MDNCF, the monocyte derived neutrophil cytotaxtic factor, which recruits neutrophils to the site of the infection.
The neutrophils reside in the blood, and travel to the capillaries, but in order to pass through the capillary wall, some modifications are necessary. The neutrophil has to change its receptors and the endophilial cells of the capillaries need to change their receptors. After this, the neutrophil can then adhere to the endophilial cell surface, where it rolls along the capillary cells until it gets to a point where it can squish in between cells called diapedesis. From there the neutrophil travels to the infectious site.
When the neutrophil encounters a bug, it takes it in by phagocytosis. The microbe contacts the membrane of the neutrophil, which envaginates to form a pocket, which then pinches off separately from the membrane to form a phagosome. This is analogous to the foreign particles of food entering your body in a separate pouch known as the stomach, so that digestion does not interfere with the internal processes of the body, or in the case of the neutrophil, so digestion does not interfere with the processes of the cell. A lysosome, filled with digestive enzymes fuses with the phagosome to make (you guessed it!) a phagolysosome, which breaks up the macromolecules into fragments, and oxidative death also occurs. The neutrophil dies in the process of killing bacteria, and then new neutrophils come along and ingest bugs and also die. In the process of dying, the neutrophil extrudes DNA, chromatin and protases, which form a neutrophil extracellular trap or NET. This keeps the bacteria from spreading and contains the bacteria. The dead neutrophil and bugs under skin make pus.
PAMPs can activate complement, which can in turn lead to the killing of pathogens in what is referred to as the alternative pathway. This can be done by a Natural Killer cell, which is also important in the innate immune response. They have a default response of "kill" when they encounter a cell. This function is so strong, they will even act against the body's best interests, and create an auto-immune response against "self." If the ligand of the Natural Killer cell is activated, unless it is turned off by the MHC class I molecule which is present on all nucleated cells, it will kill that cell, which is a problem of auto-immune disease. In many cancerous cells, the MHC class I molecule is faulty, as well as viruses can change the MHC class I molecule. Normally, it is turned off.
The next cell in after the neutrophil is the dendritic cell, which has two functions: phagocytosis and synthesis. It travels with the bacteria on its dendrites to the nearest lymph node, which in the case of the site of infection of the man's foot, happens to be in the groin. The dendritic cell links the innate immune response to the adaptive immune response, the third line of defence. The adaptive immune response is acquired and specific will the humoural aspect of the immunoglobulin mediators. It is also cell-mediated by T cells or lymphocyte effectors.
The adaptive immune response is specific to the antigen and epitope (one site of the antigen where the antibody attaches itself). The antigen presenting cells (APCs) link the antigen by means of a MHC (major histocompatibility complex) class molecule and a peptide. They may be dendritic cells, a macrophages or a B lymphocytes. The MHC molecule is unique to a specific person. There are two kinds of MHC molecules, class I and class II. Recall that class I is present on all cells in the body to turn off the Natural Killer cells. However, class II is only present on three molecules: the dendritic cells, the macrophages, and the B lymphocyte, because it is the antigen presenting molecule. After the antigen is broken down into peptides, it is presented on the dendritic cell, on the MHC class II molecule. The mjaor histocompatibility complex is called the HLA system, for Human Leukocyte Antigens, because that is where it was first seen.
Another way of phagocytosis is done by the dendritic cell. The dendritic cell goes through the same procedures as the neutrophil, except in the phagolysosome, there is the synthesis of the MHC class II molecule with an antigen fragment. The complex then comes to the surface of the cell. This holds any antigenic peptide, without specificity.
Now, before we move into the next steps of the adaptive immune response, it is necessary to take a trip back into the man's past, or more particularly, the past of his T- and B-cells. The T-cells and B-cells are made first in the yolk sac of the fetus, then in the liver and spleen after the first trimester, and finally in the bone marrow after birth. Some of these cells travel to the thymus to become T-cells. In the environment in the thymus and bone marrow, the cells mature, differentiate, and gain specificity for many possibilities of antigens.
Out of all the T-cells which mature in the thymus, only 5% leave the thymus to the blood, where as the rest of the 95% of the cells die. The vast majority of the cells produced in the thymus are anti-self, and must be eliminated to prevent a future auto-immune response. The 5% survivors have specific reactivity to only one peptide.
In the lymph nodes, the B-cells live in the portal part of the lymph node, and the T-cells are located in the madorary part of the molecule. When the dendritic cell leaves the site of infection, it travels to the site in between the B-cells and T-cells. Since the T-cell has a specific receptor that was specified in thymus pre-birth, it will see only one peptide material, one amino acid. This is the MHC peptide, which is recognized by the T-cell receptor, and thus has specificity. The CD4 molecule is involved in binding T-cells to the MHC II region of the MHC-peptide complex on the APC (antigen presenting cell). As a double-checking mechanism, the T cell synthesizes CD28, which is a co-receptor. If CD28 binds to the B7 molecule on the dendritic cell, the antibody will be produced. However, if the co-receptor is not bound, then no antibodies are made, and the organism becomes tolerant to the protein. Note that the CD28 - B7 binding is not specific.
Also, on the surface of the T-cell is a receptor called CD40L (the L stands for ligand), which is associated with an enzyme that is a ligase called CD40. The B-cell binds to the CD40L, and then the T cell produces at least three interlycukins called IL 4, 5, and 6, which cause proliferation, further maturation, and induction of the antibody. Next occurs a puzzling step where the B-cell's surface immunoglobin with antibody specificity binds to the bug on the dendrite. This is odd as the B-cell sees the whole bug, but the T-cell sees only a specific protein, yet together they make one antibody. Does this happen at the same time or in sequence? This is a mystery.
Now, both the T-cell and the B-cell have identified the bug as an immunogen, so what happens next? First, it is necessary to consider the structure of the immunogobulin antibody molecule. The molecular was initially found through electrophoresis, where the albumin molecule (a protein in blood), was seen to be homologous, but the other gobulin molecules where found to be heterogenous. The antibody activity is mostly found in the gobulin molecules, with the majority of the activity in gamma globulin and some in other classes. The immunogolbulin molecule has a tetrapeptide structure with four chains, two which are identical and heavy, and two which are identical and light. The light chains give the immunogolbulin its type, whether it is lambda, or kappa. The heavy chains determine the class of molecule: delta, my, gamma, alpha, or epsilon. Interchain and intrachain disulphide bonds are present between and within all chains.
The Fab of the immunogobulin is the antigen binding fragment, which is found at the hypervariable region, and it is divalent, so two antigens can bind. The Fc region is a crystallizable fragment which determines the biological activity of the Ig class. For example, the IgM exibits complement binding (explanation of complement forthcoming!) and is a pentameric molecule. The IgM is the first immunogobulin to respond, but it is inefficent and undergoes class switching. The IgG is present for placental transfer until the fetus can make its own immunoglobulins, is also involved with complement binding and is a dimeric molecule. Also, the IgA is involved with secretory properties including the MALT, and ensures that foreign substances that are encountered from the nasal cavity to the lungs, the digestive tract, do not cause the body to freak out. The IgE deals with mastocytophilic properties, or allergies.
After all of that information about immunoglobulins, it is important to recognize that they are often released by plasma cells, which does not sythesize immunoglobulin (antibody), but just secrete the immunoglobulins (antibodies) made during the earlier part of its B-cell life. The pro-B cell does not have surface immunoglobulin, but the pre-B cell does have this surface immunogobulin that is produced in the bone marrow, and is specific and waiting for the particular antigen to arrive in the cell. The plasma cell loses the surface immunogobulin and secretes antibody from the cell itself.
Four genes determine the structure of the heavy chain message, and each gene has multiple alleles. This is an interesting concept where randomness can actually lead to specificity. There are three regions to choose alleles from on the heavy chain: the variable region, the diversity region, and the constant region, plus an alpha, delta, mu, gamma, epsilon, determining the class. The arrangement of the immunogobulin is carried out by enzymes called the rearrangement activation genes or RAG, and the process happens in the thymus or bone marrow. This allows for hundreds of millions of different specificities, many of which may remain dormant and never be used, and others (95% of all T-cells) which may have a tendency for auto-immune response and be anti-self, so thus are knocked off by the thymus.
So, the helper T-cell has recognized the peptide linkage and the B-cell has recognized the bug, now what? Antibodies are released and then the Fab binds to the antigen, and can start the classical complement pathway (if bound to an IgM or IgG). This is a release of many protein C factors, in a complement cascade. If the factors proceed all the way to factor C9, a hole in the bacterial membrane forms, and the bug explodes. However, if the factors proceed only to C3 and C5, a so-called alternative pathway occurs, which turns on the Innate Immune Reponse, and enhances phagocytosis. Neutrophils are called in to phagocytose the bug, and then the neutrophils die releasing the net. Macrophages then arrive to clean up the pus.
In conclusion, after the immune response has finished, memory cells (either B-cells or T-cells involved in the response) are stored in the apical light zone of the lymph node. The rest of the B-cells commit suicide, called apoptosis.
Note that in the response described above, the T-cell involved was a TH2 cell, or a T Helper 2 cell. Had this not been a bacteria infection, but that of a virus, the TH1 cell would have interacted with a dendritic cell that had taken up a pathogen by means of an MHC II bound to an epitope and a TCR (T-cell receptor) of CD4+. They also have co-receptors of CD28 which binds to B7 on the dendritic cell, as a double-checking mechanism. This is exactly like the TH2 cell. But here is where the process differs: the TH1 cell releases IL-2, TNF, and INF which stimulate the Cytotoxic T-cell. The Cytotoxic T-cell's TCR which is specific for the antigen binds to the virus by recognizing an MHC class I molecule linked to the virus. This only occurs, however, if the MHC class I molecule is linked to CD8. Hence, cT cells are often described as being CD8 positive.
Once upon a time, there was a man. He may have been a good man or a bad man, I'm not entirely sure, however, the qualifier in this case is irrevelant. On one fateful day the man accidentally (or consciously, but it is highly unlikely this was the case) stepped on a piece of broken glass. The glass had shattered onto the ground, and as he was barefoot, and one of the fragments pierced his skin. http://www.youtube.com/watch?v=Rw0eIG_9tLY
Apart from the man's howl of anguish as the blood dripped down the arch of his foot, other courses of action were taking place in the man's body. Yes, his foot was throbbing and swollen, but at the cellular level, things were at work. The glass had broken through the first line of defence, the coverings of the body. The skin and the mucous membranes usually provide an unpleasant environment for the living conditions of microorganisms, but the glass had successfully penetrated the skin. Little did the man know that the glass had been infected by a bacteria, so the next line of defence had to be called into action.
The fixed tissue macrophage, present at the site of the infection, is the first cell to be involved in the innate immune response. They have toll-like receptors (TLRs), which are attracted by the PAMPs produced by the bacteria. PAMPs, pathogen-associated molecular patterns, are lipopolysaccrides or peptidylglycan molecules present on the surface of bacteria, which are absent in mammals, and are therefore considered foreign. The fixed tissue macrophage will take up some bacteria by phagocytosis, and emit a signal called the MDNCF, the monocyte derived neutrophil cytotaxtic factor, which recruits neutrophils to the site of the infection.
The neutrophils reside in the blood, and travel to the capillaries, but in order to pass through the capillary wall, some modifications are necessary. The neutrophil has to change its receptors and the endophilial cells of the capillaries need to change their receptors. After this, the neutrophil can then adhere to the endophilial cell surface, where it rolls along the capillary cells until it gets to a point where it can squish in between cells called diapedesis. From there the neutrophil travels to the infectious site.
When the neutrophil encounters a bug, it takes it in by phagocytosis. The microbe contacts the membrane of the neutrophil, which envaginates to form a pocket, which then pinches off separately from the membrane to form a phagosome. This is analogous to the foreign particles of food entering your body in a separate pouch known as the stomach, so that digestion does not interfere with the internal processes of the body, or in the case of the neutrophil, so digestion does not interfere with the processes of the cell. A lysosome, filled with digestive enzymes fuses with the phagosome to make (you guessed it!) a phagolysosome, which breaks up the macromolecules into fragments, and oxidative death also occurs. The neutrophil dies in the process of killing bacteria, and then new neutrophils come along and ingest bugs and also die. In the process of dying, the neutrophil extrudes DNA, chromatin and protases, which form a neutrophil extracellular trap or NET. This keeps the bacteria from spreading and contains the bacteria. The dead neutrophil and bugs under skin make pus.
PAMPs can activate complement, which can in turn lead to the killing of pathogens in what is referred to as the alternative pathway. This can be done by a Natural Killer cell, which is also important in the innate immune response. They have a default response of "kill" when they encounter a cell. This function is so strong, they will even act against the body's best interests, and create an auto-immune response against "self." If the ligand of the Natural Killer cell is activated, unless it is turned off by the MHC class I molecule which is present on all nucleated cells, it will kill that cell, which is a problem of auto-immune disease. In many cancerous cells, the MHC class I molecule is faulty, as well as viruses can change the MHC class I molecule. Normally, it is turned off.
The next cell in after the neutrophil is the dendritic cell, which has two functions: phagocytosis and synthesis. It travels with the bacteria on its dendrites to the nearest lymph node, which in the case of the site of infection of the man's foot, happens to be in the groin. The dendritic cell links the innate immune response to the adaptive immune response, the third line of defence. The adaptive immune response is acquired and specific will the humoural aspect of the immunoglobulin mediators. It is also cell-mediated by T cells or lymphocyte effectors.
The adaptive immune response is specific to the antigen and epitope (one site of the antigen where the antibody attaches itself). The antigen presenting cells (APCs) link the antigen by means of a MHC (major histocompatibility complex) class molecule and a peptide. They may be dendritic cells, a macrophages or a B lymphocytes. The MHC molecule is unique to a specific person. There are two kinds of MHC molecules, class I and class II. Recall that class I is present on all cells in the body to turn off the Natural Killer cells. However, class II is only present on three molecules: the dendritic cells, the macrophages, and the B lymphocyte, because it is the antigen presenting molecule. After the antigen is broken down into peptides, it is presented on the dendritic cell, on the MHC class II molecule. The mjaor histocompatibility complex is called the HLA system, for Human Leukocyte Antigens, because that is where it was first seen.
Another way of phagocytosis is done by the dendritic cell. The dendritic cell goes through the same procedures as the neutrophil, except in the phagolysosome, there is the synthesis of the MHC class II molecule with an antigen fragment. The complex then comes to the surface of the cell. This holds any antigenic peptide, without specificity.
Now, before we move into the next steps of the adaptive immune response, it is necessary to take a trip back into the man's past, or more particularly, the past of his T- and B-cells. The T-cells and B-cells are made first in the yolk sac of the fetus, then in the liver and spleen after the first trimester, and finally in the bone marrow after birth. Some of these cells travel to the thymus to become T-cells. In the environment in the thymus and bone marrow, the cells mature, differentiate, and gain specificity for many possibilities of antigens.
Out of all the T-cells which mature in the thymus, only 5% leave the thymus to the blood, where as the rest of the 95% of the cells die. The vast majority of the cells produced in the thymus are anti-self, and must be eliminated to prevent a future auto-immune response. The 5% survivors have specific reactivity to only one peptide.
In the lymph nodes, the B-cells live in the portal part of the lymph node, and the T-cells are located in the madorary part of the molecule. When the dendritic cell leaves the site of infection, it travels to the site in between the B-cells and T-cells. Since the T-cell has a specific receptor that was specified in thymus pre-birth, it will see only one peptide material, one amino acid. This is the MHC peptide, which is recognized by the T-cell receptor, and thus has specificity. The CD4 molecule is involved in binding T-cells to the MHC II region of the MHC-peptide complex on the APC (antigen presenting cell). As a double-checking mechanism, the T cell synthesizes CD28, which is a co-receptor. If CD28 binds to the B7 molecule on the dendritic cell, the antibody will be produced. However, if the co-receptor is not bound, then no antibodies are made, and the organism becomes tolerant to the protein. Note that the CD28 - B7 binding is not specific.
Also, on the surface of the T-cell is a receptor called CD40L (the L stands for ligand), which is associated with an enzyme that is a ligase called CD40. The B-cell binds to the CD40L, and then the T cell produces at least three interlycukins called IL 4, 5, and 6, which cause proliferation, further maturation, and induction of the antibody. Next occurs a puzzling step where the B-cell's surface immunoglobin with antibody specificity binds to the bug on the dendrite. This is odd as the B-cell sees the whole bug, but the T-cell sees only a specific protein, yet together they make one antibody. Does this happen at the same time or in sequence? This is a mystery.
Now, both the T-cell and the B-cell have identified the bug as an immunogen, so what happens next? First, it is necessary to consider the structure of the immunogobulin antibody molecule. The molecular was initially found through electrophoresis, where the albumin molecule (a protein in blood), was seen to be homologous, but the other gobulin molecules where found to be heterogenous. The antibody activity is mostly found in the gobulin molecules, with the majority of the activity in gamma globulin and some in other classes. The immunogolbulin molecule has a tetrapeptide structure with four chains, two which are identical and heavy, and two which are identical and light. The light chains give the immunogolbulin its type, whether it is lambda, or kappa. The heavy chains determine the class of molecule: delta, my, gamma, alpha, or epsilon. Interchain and intrachain disulphide bonds are present between and within all chains.
The Fab of the immunogobulin is the antigen binding fragment, which is found at the hypervariable region, and it is divalent, so two antigens can bind. The Fc region is a crystallizable fragment which determines the biological activity of the Ig class. For example, the IgM exibits complement binding (explanation of complement forthcoming!) and is a pentameric molecule. The IgM is the first immunogobulin to respond, but it is inefficent and undergoes class switching. The IgG is present for placental transfer until the fetus can make its own immunoglobulins, is also involved with complement binding and is a dimeric molecule. Also, the IgA is involved with secretory properties including the MALT, and ensures that foreign substances that are encountered from the nasal cavity to the lungs, the digestive tract, do not cause the body to freak out. The IgE deals with mastocytophilic properties, or allergies.
After all of that information about immunoglobulins, it is important to recognize that they are often released by plasma cells, which does not sythesize immunoglobulin (antibody), but just secrete the immunoglobulins (antibodies) made during the earlier part of its B-cell life. The pro-B cell does not have surface immunoglobulin, but the pre-B cell does have this surface immunogobulin that is produced in the bone marrow, and is specific and waiting for the particular antigen to arrive in the cell. The plasma cell loses the surface immunogobulin and secretes antibody from the cell itself.
Four genes determine the structure of the heavy chain message, and each gene has multiple alleles. This is an interesting concept where randomness can actually lead to specificity. There are three regions to choose alleles from on the heavy chain: the variable region, the diversity region, and the constant region, plus an alpha, delta, mu, gamma, epsilon, determining the class. The arrangement of the immunogobulin is carried out by enzymes called the rearrangement activation genes or RAG, and the process happens in the thymus or bone marrow. This allows for hundreds of millions of different specificities, many of which may remain dormant and never be used, and others (95% of all T-cells) which may have a tendency for auto-immune response and be anti-self, so thus are knocked off by the thymus.
So, the helper T-cell has recognized the peptide linkage and the B-cell has recognized the bug, now what? Antibodies are released and then the Fab binds to the antigen, and can start the classical complement pathway (if bound to an IgM or IgG). This is a release of many protein C factors, in a complement cascade. If the factors proceed all the way to factor C9, a hole in the bacterial membrane forms, and the bug explodes. However, if the factors proceed only to C3 and C5, a so-called alternative pathway occurs, which turns on the Innate Immune Reponse, and enhances phagocytosis. Neutrophils are called in to phagocytose the bug, and then the neutrophils die releasing the net. Macrophages then arrive to clean up the pus.
In conclusion, after the immune response has finished, memory cells (either B-cells or T-cells involved in the response) are stored in the apical light zone of the lymph node. The rest of the B-cells commit suicide, called apoptosis.
Note that in the response described above, the T-cell involved was a TH2 cell, or a T Helper 2 cell. Had this not been a bacteria infection, but that of a virus, the TH1 cell would have interacted with a dendritic cell that had taken up a pathogen by means of an MHC II bound to an epitope and a TCR (T-cell receptor) of CD4+. They also have co-receptors of CD28 which binds to B7 on the dendritic cell, as a double-checking mechanism. This is exactly like the TH2 cell. But here is where the process differs: the TH1 cell releases IL-2, TNF, and INF which stimulate the Cytotoxic T-cell. The Cytotoxic T-cell's TCR which is specific for the antigen binds to the virus by recognizing an MHC class I molecule linked to the virus. This only occurs, however, if the MHC class I molecule is linked to CD8. Hence, cT cells are often described as being CD8 positive.
Friday, 19 October 2012
Prokaryotic Gene Expression
Transcriptional Control
The major mechanism for controlling protein production in the cell is determined by which genes are transcribed to encode a particular protein. This is important as the structure and function of the cell is determined by the proteins it contains. The cell regulates the proteins and speed of which they are produced by repressing and activating the gene in question.
repressing a gene: the corresponding mRNA is transcribed at a low rate (meaning little to nothing in the cell)
activating a gene: the corresponding mRNA is transcribed at a much higher rate (meaning up to 1000x or more RNA is transcribed)
Purpose of Transcriptional Control...
...in Single-Celled Organisms
Transcription of genes is regulated to adjust to changes in the nutritional and physical environment. The cell will produce only the proteins required for survival and will proliferate under the particular environmental conditions it experiences.
... in Multicellular Organisms
Transcription of genes is regulated to ensure coordination during embryonic development and tissue differentiation. Again, the level of organization is more complex.
Operons are Efficient for Control
Recall that operons are sequences that encode enzymes in a row that are involved in a particular metabolic pathway or proteins that interact to form a large multi-unit protein. This is characteristic of procaryotes, as eukaryotes have their proteins in different regions of the chromosome separated by large uncoded regions called introns. Point being, in E coli, half of the genes are clustered into such operons, with the trp operon coding for five enzymes needed in the biosynthesis of tryptophan, and the lac operon coding for three enzymes needed in the biosynthesis of lactose.
All genes on an operon are coordinately regulated, meaning that they are repressed and activated to the same extent, which is efficient and economic for controlling transcription regulation in bacteria. The regulation happens through RNA polymerase and specific repressor and activator proteins. To initate transcription, RNA polymerase must associate with a sigma factor, most commonly  . These sigma factors are not conserved in eukaryotes.
. These sigma factors are not conserved in eukaryotes.
Function of Sigma Factors
Sigma Factors recognize specific DNA sequences as promoters and recruit RNA polymerase which is a lot simpler sounding than that whole eukaryotic preinitiation complex razzmatazz. After transcription is initiated the sigma factor peaces out, or is released from the promoter upstream of the start site. recognizes the sequence TTGACA (in the -30 region) ...15-17 bps... TATAAT (in the -10 region). On the other hand,  recognizes a very difference sequence as it is involved with metabolizing nitrogen. Eg: the regions on the DNA which these sigma factors recognize are diverse.
recognizes a very difference sequence as it is involved with metabolizing nitrogen. Eg: the regions on the DNA which these sigma factors recognize are diverse.
The E coli Lac Operon Saga
The E coli lac operon exists in a repressed state.
The E coli Lac Operon encodes the three enzymes required to metabolize lactose in the cell. However, the cell much prefers to get its energy from glucose, so the enzymes will only be transcribed if there is a) high lactose and b) low glucose in the cell. Normally, this is not the case, so the lac repressor binds to the operator, blocking the start site, and the promoter is ready for polymerase to bind, no transcription occurs.
De-repressing the lac operon.
However, when lactose enters the cell in high qualities, it binds to the lac repressor, causing a conformational change and for it to release from the operon. Thenbinds and recruits the polymerase to the promoter. Transcription occurs, until lactose levels are low once more in the cell, and then the lac repressor will again bind to the lac operator. It should be mentioned though, that transcription levels of the proteins are low for this scenario as glucose (the preferred metabolite) is still present in the cell.
Activating the lac operon.
If both glucose levels are low in the cell, and lactose levels are high, then something else happens with the operon. cAMP is produced by the low glucose levels in the cell and then binds to CAP (catabolite activator protein) from there, the complex binds to a site upstream of the promoter called the CAP site. The lactose binds to the lac repressor, causing the conformational change and release from the operator, then the polymerase binds to the promoter complexed with. The cAMP and CAP complex greatly stimulate the rate of transcription.
The major mechanism for controlling protein production in the cell is determined by which genes are transcribed to encode a particular protein. This is important as the structure and function of the cell is determined by the proteins it contains. The cell regulates the proteins and speed of which they are produced by repressing and activating the gene in question.
repressing a gene: the corresponding mRNA is transcribed at a low rate (meaning little to nothing in the cell)
activating a gene: the corresponding mRNA is transcribed at a much higher rate (meaning up to 1000x or more RNA is transcribed)
Purpose of Transcriptional Control...
...in Single-Celled Organisms
Transcription of genes is regulated to adjust to changes in the nutritional and physical environment. The cell will produce only the proteins required for survival and will proliferate under the particular environmental conditions it experiences.
... in Multicellular Organisms
Transcription of genes is regulated to ensure coordination during embryonic development and tissue differentiation. Again, the level of organization is more complex.
Operons are Efficient for Control
Recall that operons are sequences that encode enzymes in a row that are involved in a particular metabolic pathway or proteins that interact to form a large multi-unit protein. This is characteristic of procaryotes, as eukaryotes have their proteins in different regions of the chromosome separated by large uncoded regions called introns. Point being, in E coli, half of the genes are clustered into such operons, with the trp operon coding for five enzymes needed in the biosynthesis of tryptophan, and the lac operon coding for three enzymes needed in the biosynthesis of lactose.
. These sigma factors are not conserved in eukaryotes.Function of Sigma Factors
recognizes the sequence TTGACA (in the -30 region) ...15-17 bps... TATAAT (in the -10 region). On the other hand, recognizes a very difference sequence as it is involved with metabolizing nitrogen. Eg: the regions on the DNA which these sigma factors recognize are diverse.The E coli Lac Operon Saga
The E coli lac operon exists in a repressed state.
The E coli Lac Operon encodes the three enzymes required to metabolize lactose in the cell. However, the cell much prefers to get its energy from glucose, so the enzymes will only be transcribed if there is a) high lactose and b) low glucose in the cell. Normally, this is not the case, so the lac repressor binds to the operator, blocking the start site, and the promoter is ready for polymerase to bind, no transcription occurs.
De-repressing the lac operon.
However, when lactose enters the cell in high qualities, it binds to the lac repressor, causing a conformational change and for it to release from the operon. Then
binds and recruits the polymerase to the promoter. Transcription occurs, until lactose levels are low once more in the cell, and then the lac repressor will again bind to the lac operator. It should be mentioned though, that transcription levels of the proteins are low for this scenario as glucose (the preferred metabolite) is still present in the cell.Activating the lac operon.
If both glucose levels are low in the cell, and lactose levels are high, then something else happens with the operon. cAMP is produced by the low glucose levels in the cell and then binds to CAP (catabolite activator protein) from there, the complex binds to a site upstream of the promoter called the CAP site. The lactose binds to the lac repressor, causing the conformational change and release from the operator, then the polymerase binds to the promoter complexed with
. The cAMP and CAP complex greatly stimulate the rate of transcription.Pol II Preinitiation Complex (Pol II and Company)
In order to initiate transcription, general transcription factors are required to place Pol II at the transcription start site, which is a promoter containing a TATA box. They also help separate DNA strands so that the template strand can enter the active site of the enzyme. As these initiation factors are necessary for most, if not all, promoters transcribed by RNA Polymerase II, they are called general transcription factors. Recall that transcription factors are proteins. Together, these transcription factors form what is referred to as a preinitiation complex in vivo and in vitro. We are going to explain the in vitro preinitiation complex and the steps of the order of various transcription factor binding.
1) The first transcription factor to bind to the TATA box, is not shockingly, the TATA box binding protein, or TBP. It folds into a saddle-like structure, because of its C-terminal domain, and from there, it interacts with the minor groove of DNA. It bends the helix significantly.
As the DNA-binding surface of the TBP is highly conserved in eukaryotes, it helps explain the high conservation of the TATA box promoter element.
2) Next, TFIIB binds by its C-terminal domain making contact with both TBP and either side of the TATA box. The N-terminal domain is inserted into the RNA exit channel of Pol II and helps Pol II in melting the DNA strands at the transcription start site. It also interacts with the template strand near the Pol II active site.
3) TFIIF (heterodimer) and Pol II bind, which positions Pol II over the start site.
4) After, that TFIIE (tetrameric with two different subunits) binds next to TFIIF and PolII on the side closest to the upstream direction. This creates a docking site for TFIIH.
5) Now, TFIIH (10 different subunits) binds, completing the assembly of the Pol II Preinitiation Complex.
6) Helicase activity of one of the TFIIH subunits uses energy from ATP hydrolysis to help unwind the DNA duplex at the start site, which allows Pol II to form an open complex (transcription bubble). There is a release of the general transcription factors, except for TBP. Pol II elongates the nascent mRNA strand, and has a phosphorylated CTD, which allows for association of the enzymes required to add the 5' cap.
Brief note of some differences in vivo:
In vivo, TBP is part of TFIID, which is a complex of not only TBP, but also 13 other subunits called TAFs. Also, TFIIA is required to form the preinitiation complex and binds to TBP and the TATA box.
1) The first transcription factor to bind to the TATA box, is not shockingly, the TATA box binding protein, or TBP. It folds into a saddle-like structure, because of its C-terminal domain, and from there, it interacts with the minor groove of DNA. It bends the helix significantly.
As the DNA-binding surface of the TBP is highly conserved in eukaryotes, it helps explain the high conservation of the TATA box promoter element.
2) Next, TFIIB binds by its C-terminal domain making contact with both TBP and either side of the TATA box. The N-terminal domain is inserted into the RNA exit channel of Pol II and helps Pol II in melting the DNA strands at the transcription start site. It also interacts with the template strand near the Pol II active site.
3) TFIIF (heterodimer) and Pol II bind, which positions Pol II over the start site.
4) After, that TFIIE (tetrameric with two different subunits) binds next to TFIIF and PolII on the side closest to the upstream direction. This creates a docking site for TFIIH.
5) Now, TFIIH (10 different subunits) binds, completing the assembly of the Pol II Preinitiation Complex.
6) Helicase activity of one of the TFIIH subunits uses energy from ATP hydrolysis to help unwind the DNA duplex at the start site, which allows Pol II to form an open complex (transcription bubble). There is a release of the general transcription factors, except for TBP. Pol II elongates the nascent mRNA strand, and has a phosphorylated CTD, which allows for association of the enzymes required to add the 5' cap.
Brief note of some differences in vivo:
In vivo, TBP is part of TFIID, which is a complex of not only TBP, but also 13 other subunits called TAFs. Also, TFIIA is required to form the preinitiation complex and binds to TBP and the TATA box.
Thursday, 18 October 2012
Pol II and Friends
Finding Pol I, II, III
When scientists found the eukaryotic RNA Polymerases I II, and III, they did so by separating the proteins by chromatography. A poison from a mushroom called a-amantin was passed with a protein extract from the nuclei of cultured eukaryotic cells through a DEAE Sephadex columun. After the protein was collected at various different times, it was eluted (washed) with a solution of constantly increasing NaCl. Fractions from this elute were assayed for RNA polymerase activity (is the RNA synthesized or not?), and it was found that where Pol I was insenitive to a-amantin at both 1 umg and and 10 umg, Pol II was sensitive to a-amantin at 1 umg, and Pol II was intermediately sensitive to a-amantin, meaning insensitive at 1 umg, but sensitive at a higher concentration of 10 umg.
Conservation in Polymerase Structures
It isn't too surprising that there are highly conserved structures between bacterial E coli RNA polymerase (bacteria) and Yeast RNA polymerase (eukaryotes). After all, transcribing RNA from DNA is pretty important to life, and conservation indicates the polymerase enzyme was around early on in its evolutionary history. So not only does yeast RNA polymerase share similar major structures similar to the B and B' E coli RNAs, but also possesses a w-like and two nonidentical a-like subunits.
However, the eukaryotic RNA polymerases are more complex than the bacterial counterparts. All three yeast RNA polymerases contain four additional subunits common to them, but not shared with prokaryotes.
Also each eukaryotic nuclear RNA polymerase has several enzyme specific subunits that are not present in the other two RNA polymerases. Three of these additional subunits of Pol I and Pol III are homologous to three of the additional subunits of Pol II. The other two Pol I-specific subunits are homologous to Pol II transcription factor TFIIF. The four additional subunits of Pol III are homologous to the Pol II transcription factors TFIIF and TFIIE. So in conclusion, Pol II has three additional specific subunits, Pol I has 5, and Pol III has 7.
* Structure of Yeast RNA Polymerase
I may go back to this post and add more details about the Yeast RNA Polymerase domains, but as of now, I am unsure if this level of intricacy is required. If I do, you will see an additional paragraph section deal added tomorrow, if not, this little blurb will be deleted.
Carboxyl-Terminal Domain (CTD)
This is the largest subunit on the RNA Polymerase II, which contributes to the vitality of the organism. Without a certain number of repeats (10 in yeast), the polymerase will not function and the organism will die. The stretch of 7 amino acid is nearly always precisely repeated, and is Tyr-Ser-Pro-Ser-Thr-Pro-Ser in mammals. A cute way to remember this sequence is the statement "this subject proves that some people study"; sorry it is kind of negative, but the best I could come up with under strain. Yeast had 26 or more of these repeats, where as vertebrates have 523.
RNA pol II molecules that first initiate transcription have an unphosphorylated tail, but once the polymerase initiates transcription and begins to move away from the promoter, many of the serine and some tryosine residues in the CTD are phosphorylated.
This phenomenon can be seen in vivo in Drosphilia staining. A puffed chromosomal region is stained by antibodies specific for phosphorylation (red) or unphosphorylated (green) from the salivary glands.
Now, as an aside, I've never been able to stain Drosphilia salivary glands successfully. I decapitated my larvae three or four times with painful lack of precision, and when asking a TA for assistance, was informed that no one knows how to make the puncture wound to spew out the desired contents under the dissection microscope. I might be just a little sore about this failing, as three to four Drosphilia died needlessly in infancy and did not make it to the next test cross where they eventually achieved the honour of death by morgue jar. Anyway, my lab partner was kind of squeamish about the process, and out of a class of thirty only one pair managed to find the classic five stranded chromosome. I digress. But anyone who can stain a chromosome with two types of antibodies, get the bally chromosome from larvae guts in the first place, and see the regions of interest, is in my opinion, kind of a boss. Kudos to them.
When scientists found the eukaryotic RNA Polymerases I II, and III, they did so by separating the proteins by chromatography. A poison from a mushroom called a-amantin was passed with a protein extract from the nuclei of cultured eukaryotic cells through a DEAE Sephadex columun. After the protein was collected at various different times, it was eluted (washed) with a solution of constantly increasing NaCl. Fractions from this elute were assayed for RNA polymerase activity (is the RNA synthesized or not?), and it was found that where Pol I was insenitive to a-amantin at both 1 umg and and 10 umg, Pol II was sensitive to a-amantin at 1 umg, and Pol II was intermediately sensitive to a-amantin, meaning insensitive at 1 umg, but sensitive at a higher concentration of 10 umg.
Introduction to the Three Eukaryotic Polymerases
Polymerase
|
RNA Transcribed
|
RNA Function
|
Polymerase I
|
Pre r-RNA (28S, 18S, 5.8S rRNAs)
|
ribosome complements, protein synthesis
|
Polymerase II
|
mRNA
snRNA
siRNA
miRNAs
|
encodes protein
RNA splicing
chromatin-mediated repression translational control
translation control
|
Polymerase III
|
tRNA
5S rRNA
snRNA U6
7S RNA
other stable short RNAs
|
protein synthesis
ribosome component, protein synthesis
RNA splicing
signal-recognition particle for insertion of polypeptides into the ER
various functions, most unknown
|
Conservation in Polymerase Structures
It isn't too surprising that there are highly conserved structures between bacterial E coli RNA polymerase (bacteria) and Yeast RNA polymerase (eukaryotes). After all, transcribing RNA from DNA is pretty important to life, and conservation indicates the polymerase enzyme was around early on in its evolutionary history. So not only does yeast RNA polymerase share similar major structures similar to the B and B' E coli RNAs, but also possesses a w-like and two nonidentical a-like subunits.
However, the eukaryotic RNA polymerases are more complex than the bacterial counterparts. All three yeast RNA polymerases contain four additional subunits common to them, but not shared with prokaryotes.
Also each eukaryotic nuclear RNA polymerase has several enzyme specific subunits that are not present in the other two RNA polymerases. Three of these additional subunits of Pol I and Pol III are homologous to three of the additional subunits of Pol II. The other two Pol I-specific subunits are homologous to Pol II transcription factor TFIIF. The four additional subunits of Pol III are homologous to the Pol II transcription factors TFIIF and TFIIE. So in conclusion, Pol II has three additional specific subunits, Pol I has 5, and Pol III has 7.
* Structure of Yeast RNA Polymerase
I may go back to this post and add more details about the Yeast RNA Polymerase domains, but as of now, I am unsure if this level of intricacy is required. If I do, you will see an additional paragraph section deal added tomorrow, if not, this little blurb will be deleted.
Carboxyl-Terminal Domain (CTD)
This is the largest subunit on the RNA Polymerase II, which contributes to the vitality of the organism. Without a certain number of repeats (10 in yeast), the polymerase will not function and the organism will die. The stretch of 7 amino acid is nearly always precisely repeated, and is Tyr-Ser-Pro-Ser-Thr-Pro-Ser in mammals. A cute way to remember this sequence is the statement "this subject proves that some people study"; sorry it is kind of negative, but the best I could come up with under strain. Yeast had 26 or more of these repeats, where as vertebrates have 523.
RNA pol II molecules that first initiate transcription have an unphosphorylated tail, but once the polymerase initiates transcription and begins to move away from the promoter, many of the serine and some tryosine residues in the CTD are phosphorylated.
This phenomenon can be seen in vivo in Drosphilia staining. A puffed chromosomal region is stained by antibodies specific for phosphorylation (red) or unphosphorylated (green) from the salivary glands.
Now, as an aside, I've never been able to stain Drosphilia salivary glands successfully. I decapitated my larvae three or four times with painful lack of precision, and when asking a TA for assistance, was informed that no one knows how to make the puncture wound to spew out the desired contents under the dissection microscope. I might be just a little sore about this failing, as three to four Drosphilia died needlessly in infancy and did not make it to the next test cross where they eventually achieved the honour of death by morgue jar. Anyway, my lab partner was kind of squeamish about the process, and out of a class of thirty only one pair managed to find the classic five stranded chromosome. I digress. But anyone who can stain a chromosome with two types of antibodies, get the bally chromosome from larvae guts in the first place, and see the regions of interest, is in my opinion, kind of a boss. Kudos to them.
Wednesday, 17 October 2012
Gene Organisation: Prokaryotes verus Eukaryotes
Prokaryotes
In bacteria, the genes that encode proteins that serve a particular function together are found in a contiguous (sharing a common border) array in the DNA. These arrangements are called operons. The operons form a single unit and will share a promoter, and when transcribed, will produce a single strand of mRNA. From there, the various proteins will be translated, and thus, results in the coordinate expression of all of the genes in the operon. This means that all of the genes in the operon are transcribed and translated. Note that in prokaryotes, very few gaps of noncoding DNA are found, and so mRNA is transcribed directly from the get-go. Also, as DNA is not sequestered in a nucleus, as prokaryotes do not have organelles, translation starts by ribosomes at the 3' end of the nascent mRNA while the polymerase is still attached and carrying out transcription. Such processes are known as dynamic.
Eukaryotes
As eukaryotes are more complex, genes are not organized into operons that code for proteins that serve a particular function. In fact, the genes are physically separated in the DNA, and are located on different chromosomes for instance. Co-regulation in eukaryotes is not achieved simply by physical linkage like in prokaryotes. Instead, each gene is transcribed from its own promoter, which is translated to form a single polypeptide. This contrasts with prokaryotes which from various polypeptides from the same operon. Also the protein-coding sequence of mRNA is discontinuous with the template DNA strand. How is this possible? After all, the mRNA was transcribed directly from this strand, so they should contain identical information. Not so. The eukaryotic gene contains pieces of coding sequence called exons, separated by pieces of noncoding sequence called introns. During mRNA processing, the introns are removed from the initial primary transcript (the RNA copy of the entire DNA sequence).
Eukaryotic mRNA Processing
Since transcription occurs in the nucleus, for eukaryotes, no translation can occur until the mRNA is processed. First, the DNA is transcribed to form a pre-cursor mRNA, also known as the primary transcript. This long sequence contains all of the information of the original DNA strand.
As soon as the 5' end of the RNA emerges from the polymerase, it is given a 5' cap, a 7-methylguanylate which identifies it as an mRNA so it will eventually be exported to the cytoplasm and stabilizes the structure from being degraded by other enzymes. The capping is done by enzymes and it is connected by 5' 5' triphosphate linkage.
Then, the primary transcript is cleaved at a 3' end site to yield a free 3' hydroxyl group to which adelylinic acid residues (100-250) are added to form a poly(A) tail. This is done by poly(A) polymerase.
Finally, RNA splicing occurs, where the transcript is cleaved and the introns are removed (intron excision), and the exons are connected together again (exon ligation).
Eukatryotic mRNAs contain untranslated regions or (UTRs) at both ends, with the 5' UTR having around 100 nucleotides and the 3' UTR having around several kilobases. Bacteria also have UTRs, but they are smaller, around 10 nucleotides only.
Alternative RNA Splicing
Alternative RNA splicing leads to greater diversity in the types of proteins encoded by the genomes of higher organisms. How this works is a single gene can have exons spliced out of its sequence giving rise to different forms of a protein called isoforms that are encoded by the same gene.
For example, fibronectin, a multidomain protein found in mammals, is processed by alternative splicing. In fibroblasts, the fibronectin mRNAs contain exons EIIIA and EIIIB, which then encode amino acid sequences that bind tightly to proteins in the fibroblast plasma membrane. In heptatocytes (liver cells), mRNAs do not contain exons EIIIA and EIIIB, as they have been excised from the mRNA sequence. Thus, fibronectin produced by the liver, does not adhere to proteins. More than 20 different kinds of fibronectin exist; all produced by alternative splicing.
In bacteria, the genes that encode proteins that serve a particular function together are found in a contiguous (sharing a common border) array in the DNA. These arrangements are called operons. The operons form a single unit and will share a promoter, and when transcribed, will produce a single strand of mRNA. From there, the various proteins will be translated, and thus, results in the coordinate expression of all of the genes in the operon. This means that all of the genes in the operon are transcribed and translated. Note that in prokaryotes, very few gaps of noncoding DNA are found, and so mRNA is transcribed directly from the get-go. Also, as DNA is not sequestered in a nucleus, as prokaryotes do not have organelles, translation starts by ribosomes at the 3' end of the nascent mRNA while the polymerase is still attached and carrying out transcription. Such processes are known as dynamic.
Eukaryotes
As eukaryotes are more complex, genes are not organized into operons that code for proteins that serve a particular function. In fact, the genes are physically separated in the DNA, and are located on different chromosomes for instance. Co-regulation in eukaryotes is not achieved simply by physical linkage like in prokaryotes. Instead, each gene is transcribed from its own promoter, which is translated to form a single polypeptide. This contrasts with prokaryotes which from various polypeptides from the same operon. Also the protein-coding sequence of mRNA is discontinuous with the template DNA strand. How is this possible? After all, the mRNA was transcribed directly from this strand, so they should contain identical information. Not so. The eukaryotic gene contains pieces of coding sequence called exons, separated by pieces of noncoding sequence called introns. During mRNA processing, the introns are removed from the initial primary transcript (the RNA copy of the entire DNA sequence).
Eukaryotic mRNA Processing
Since transcription occurs in the nucleus, for eukaryotes, no translation can occur until the mRNA is processed. First, the DNA is transcribed to form a pre-cursor mRNA, also known as the primary transcript. This long sequence contains all of the information of the original DNA strand.
As soon as the 5' end of the RNA emerges from the polymerase, it is given a 5' cap, a 7-methylguanylate which identifies it as an mRNA so it will eventually be exported to the cytoplasm and stabilizes the structure from being degraded by other enzymes. The capping is done by enzymes and it is connected by 5' 5' triphosphate linkage.
Then, the primary transcript is cleaved at a 3' end site to yield a free 3' hydroxyl group to which adelylinic acid residues (100-250) are added to form a poly(A) tail. This is done by poly(A) polymerase.
Finally, RNA splicing occurs, where the transcript is cleaved and the introns are removed (intron excision), and the exons are connected together again (exon ligation).
Eukatryotic mRNAs contain untranslated regions or (UTRs) at both ends, with the 5' UTR having around 100 nucleotides and the 3' UTR having around several kilobases. Bacteria also have UTRs, but they are smaller, around 10 nucleotides only.
Alternative RNA Splicing
Alternative RNA splicing leads to greater diversity in the types of proteins encoded by the genomes of higher organisms. How this works is a single gene can have exons spliced out of its sequence giving rise to different forms of a protein called isoforms that are encoded by the same gene.
For example, fibronectin, a multidomain protein found in mammals, is processed by alternative splicing. In fibroblasts, the fibronectin mRNAs contain exons EIIIA and EIIIB, which then encode amino acid sequences that bind tightly to proteins in the fibroblast plasma membrane. In heptatocytes (liver cells), mRNAs do not contain exons EIIIA and EIIIB, as they have been excised from the mRNA sequence. Thus, fibronectin produced by the liver, does not adhere to proteins. More than 20 different kinds of fibronectin exist; all produced by alternative splicing.
Tuesday, 16 October 2012
Transcription: A Starting Point
Transcription in Brief
Transcription is when the DNA strand is copied or transcribed into the four-base language of RNA.
This may seem a trivial statement, however when things can get complicated, it is an important statement to return to, as it provides the bigger picture of the significance of what is going on in the cell. (or test tube if you go all in vitro and such)
However, the details about transcription are anything but brief. For this post I will be covering the basic outline of transcription and from there, future posts will probably involve more precise, involved descriptions about the process.
Overview of Transcription
So, let's return to the DNA, sitting in its double helix form in the nucleus. During transcription one of its strands will act as a template, and will be denatured locally to allow access for an incoming ribonucleotide triphosphates (rNTPs). These rNTPs can be thought of as the building blocks of RNA, and bind to the complementary DNA bases with Watson-Crick base pairing. Remember however, that RNA uses uracil instead of thymine. RNA polymerase catalyzes a polymerase reaction to join the rNTPs from 5' to 3'. This is opposite in directionality of the template DNA strand, which goes from 3' to 5'. The sequence the RNA is transcribing is then the same as the nontemplate DNA strand.
What is polymerization?
The mechanism works via a nucleophilic attack, where a nucleophile is a species that can donate an electron pair. In this case, the nucleophile is the 3' oxygen of the growing RNA chain, and it attacks the alpha phosphate, which is the first phosphate attached to the ribose of a chain of three of the incoming nucleotide that will be the next in the RNA sequence. This results in the formation of a phosphodiester bond, and the release of a phosphate (PPi). Again, the rNTPs are added from 5' to 3'.
Why is polymerization favoured?
The high-energy bond between the alpha and beta phosphates of the incoming nucleotide (the first phosphate attached to the ribose, and the one next door) is replaced by a lower-energy phosphodiester bond between nucleotides. Also, another enzyme called pyrophosphatase, catalyzes cleavage of the released PPi into two moleules of inorganic phosphate, and pushes the equilibrium to favour chain elongation. In other words, the pyrophosphatase makes it easier for the phosphate to leave the high energy bond.
Some Conventions When Discussing Transcription
The site on the DNA where the RNA polymerase begins transcription is labelled +1, called the start site.
Downstream = the direction of transcription (+ from the start site)
Upstream = the direction opposite transcription (- from the start site)
Stages in Transcription
Initiation
1) Polymerase binds to promoter sequence in the duplex DNA with the aid of several general transcription factors (proteins). This is a "Closed Complex" as the DNA is still double stranded.
2) Polymerase melts duplex DNA near transcription start site for 12-14 base pairs, forming a transcription bubble, which seperates the DNA strands to allow rNTPs access to the DNA template strand sequence. It also allows the template strand to have access to the active site of polymerase so that it can catalyzes phosphodiester linkage. Now the DNA is called "Open Complex."
3) Polymerase catalyzes phosphodiester linnkage of two initial rNTPs.
Elongation
4) To undertake the elongation journey, polymerase dissociates from the promoter sequence and general transcription factors. Polymerase advances 3' to 5' down template strand, melting duplex DNA and adding RNTPs to growing RNA, while guiding the DNA strands so they hybridize at the upstream end of the transcription bubble. The transcription bubble has a melted region of ~14 base pairs of which ~8 nucleotides at the 3' end of the growing RNA strand remain base-paired to the template DNA strand. The elongation complex consists of RNA polymerase, template DNA, and growing/nascent RNA. The speed of elongation is around 1000 nucleotides a minute.
Termination
5) At transcription stop site, polymerase releases completed RNA and disassociates from DNA. Once the polymerase is dissociated it is good to go find more DNAs and transcribe some more. It is not specific to a certain gene.
Structure of RNA Polymerases
Structure of RNA polymerases is conserved between bacteria, archaea, and eukaryotic cells.
Bacteria RNA polymerase: 2 related large subunits (B and B'), 2 smaller identical subunits (a), and 1 copy of 5th subunit (w)
The w subunit stabilizes the molecule, no aid to transcription.
DNA bends sharply when entering the RNA Polymerase molecule.
Transcription is when the DNA strand is copied or transcribed into the four-base language of RNA.
This may seem a trivial statement, however when things can get complicated, it is an important statement to return to, as it provides the bigger picture of the significance of what is going on in the cell. (or test tube if you go all in vitro and such)
However, the details about transcription are anything but brief. For this post I will be covering the basic outline of transcription and from there, future posts will probably involve more precise, involved descriptions about the process.
Overview of Transcription
So, let's return to the DNA, sitting in its double helix form in the nucleus. During transcription one of its strands will act as a template, and will be denatured locally to allow access for an incoming ribonucleotide triphosphates (rNTPs). These rNTPs can be thought of as the building blocks of RNA, and bind to the complementary DNA bases with Watson-Crick base pairing. Remember however, that RNA uses uracil instead of thymine. RNA polymerase catalyzes a polymerase reaction to join the rNTPs from 5' to 3'. This is opposite in directionality of the template DNA strand, which goes from 3' to 5'. The sequence the RNA is transcribing is then the same as the nontemplate DNA strand.
What is polymerization?
The mechanism works via a nucleophilic attack, where a nucleophile is a species that can donate an electron pair. In this case, the nucleophile is the 3' oxygen of the growing RNA chain, and it attacks the alpha phosphate, which is the first phosphate attached to the ribose of a chain of three of the incoming nucleotide that will be the next in the RNA sequence. This results in the formation of a phosphodiester bond, and the release of a phosphate (PPi). Again, the rNTPs are added from 5' to 3'.
Why is polymerization favoured?
The high-energy bond between the alpha and beta phosphates of the incoming nucleotide (the first phosphate attached to the ribose, and the one next door) is replaced by a lower-energy phosphodiester bond between nucleotides. Also, another enzyme called pyrophosphatase, catalyzes cleavage of the released PPi into two moleules of inorganic phosphate, and pushes the equilibrium to favour chain elongation. In other words, the pyrophosphatase makes it easier for the phosphate to leave the high energy bond.
Some Conventions When Discussing Transcription
The site on the DNA where the RNA polymerase begins transcription is labelled +1, called the start site.
Downstream = the direction of transcription (+ from the start site)
Upstream = the direction opposite transcription (- from the start site)
Stages in Transcription
Initiation
1) Polymerase binds to promoter sequence in the duplex DNA with the aid of several general transcription factors (proteins). This is a "Closed Complex" as the DNA is still double stranded.
2) Polymerase melts duplex DNA near transcription start site for 12-14 base pairs, forming a transcription bubble, which seperates the DNA strands to allow rNTPs access to the DNA template strand sequence. It also allows the template strand to have access to the active site of polymerase so that it can catalyzes phosphodiester linkage. Now the DNA is called "Open Complex."
3) Polymerase catalyzes phosphodiester linnkage of two initial rNTPs.
Elongation
4) To undertake the elongation journey, polymerase dissociates from the promoter sequence and general transcription factors. Polymerase advances 3' to 5' down template strand, melting duplex DNA and adding RNTPs to growing RNA, while guiding the DNA strands so they hybridize at the upstream end of the transcription bubble. The transcription bubble has a melted region of ~14 base pairs of which ~8 nucleotides at the 3' end of the growing RNA strand remain base-paired to the template DNA strand. The elongation complex consists of RNA polymerase, template DNA, and growing/nascent RNA. The speed of elongation is around 1000 nucleotides a minute.
Termination
5) At transcription stop site, polymerase releases completed RNA and disassociates from DNA. Once the polymerase is dissociated it is good to go find more DNAs and transcribe some more. It is not specific to a certain gene.
Structure of RNA Polymerases
Structure of RNA polymerases is conserved between bacteria, archaea, and eukaryotic cells.
Bacteria RNA polymerase: 2 related large subunits (B and B'), 2 smaller identical subunits (a), and 1 copy of 5th subunit (w)
The w subunit stabilizes the molecule, no aid to transcription.
DNA bends sharply when entering the RNA Polymerase molecule.
Tuesday, 9 October 2012
RNA Binding Domains and hnRNPs
RNA Binding Domains
The RNA binding domains are present on population of proteins which associate with RNA during different processes. They were discovered through HnRNPs, heterogeneous ribonucleoprotein particles, which associate with pre-mRNAs by examining their conservative features. Like DNA binding domains, the proteins' RNA binding domains have several structural motifs.
RNA Recognition Motif (RRM)
The most common RNA binding domain. 80 amino acids fold into a 4-stranded B sheet flanked by 2 a-helices, which contain RNP1 and RNP2 motifs that contact the phosphates of RNA. This is a charged interaction as the negative phosphate backbone attracts a positively charged binding motif.
RRG Box
The motif contains 5 Arg-Gly-Gly repeats interspersed with aromatic amino acids, such as Phe, Tyr, and Trp. The structure is unknown. It is a charged interaction.
KH Motif
This has 35 residues, with a similar structure to the RRM domain, but the RNA binds by interacting with a hydrophobic surface formed by the alpha helices and one beta strand.
A Note about hnRNPs
hnRNPs are a set of proteins that contain hnRNAs, which are pre-mRNAs and RNA processing intermediates with introns still present in their sequences. They prevent undue interaction from occurring between the pre-mRNA with other components of the cell as well as prevent it from interacting with itself to form a secondary structure, which would muck things up. Also, they are involved with different steps of RNA processing, sometimes associating with the splicing apparatus for instance, and transporting the mRNA from the nucleus to the cytoplasm.
Post-Transcription: Capping the 5' End of mRNA
So this is a continuation of the transcription saga and consists of the plot after transcription occurs.
Eukaryotic mRNA Processing
There are four important steps regarding post-transcriptional processing of mRNA:
1) Transcription and 5' Capping of mRNA
2) Cleavage at the Poly(A) site
3) Polyadenylation
4) RNA splicing
Capping the 5' End of the mRNA
The mRNA is the primary RNA transcript; the sequence coded from the template DNA strand. To prepare the mRNA to become a fully functioning polypeptide, the introns have to be removed. However, before this happens, the first step that occurs is the capping of the 5' end of the mRNA. Why? Well, capping the mRNA protects it from degradation from nucleases. Exonucleuses recognize the free end of the mRNA of the OH group, so to make this OH group invisible, it must be capped. 7-methylguanosine is added to the 5' end when the transcript is 25-30 nucleotides long even while its still associated with RNA Pol II. This is a dynamic process, so transcription can still be in session while the mRNA is processed. The dimeric capping enzyme associates with the unstructured CTD tail of RNA Pol II, becomes phosphorylated and drags along with it the enzymes required to process the mRNA. One subunit of the capping enzyme removes the gamma-phosphate from the 5' end of the RNA and the other converts GMP from GTP to the 5' diphosphate of the transcript.
Eukaryotic mRNA Processing
There are four important steps regarding post-transcriptional processing of mRNA:
1) Transcription and 5' Capping of mRNA
2) Cleavage at the Poly(A) site
3) Polyadenylation
4) RNA splicing
Capping the 5' End of the mRNA
The mRNA is the primary RNA transcript; the sequence coded from the template DNA strand. To prepare the mRNA to become a fully functioning polypeptide, the introns have to be removed. However, before this happens, the first step that occurs is the capping of the 5' end of the mRNA. Why? Well, capping the mRNA protects it from degradation from nucleases. Exonucleuses recognize the free end of the mRNA of the OH group, so to make this OH group invisible, it must be capped. 7-methylguanosine is added to the 5' end when the transcript is 25-30 nucleotides long even while its still associated with RNA Pol II. This is a dynamic process, so transcription can still be in session while the mRNA is processed. The dimeric capping enzyme associates with the unstructured CTD tail of RNA Pol II, becomes phosphorylated and drags along with it the enzymes required to process the mRNA. One subunit of the capping enzyme removes the gamma-phosphate from the 5' end of the RNA and the other converts GMP from GTP to the 5' diphosphate of the transcript.
Monday, 8 October 2012
Activation Domains
The activation domains are responsible for interacting with other proteins. They are unstructured until bound by a ligand, and then they can interact with other proteins. Less sequence consensus is required for activation domains than for DNA-binding domains. Many have a high percentage of one or two particular amino acids (Asp, Glu, Gln, Pro, Ser, Thr). Acidic activation domains, which are those with Aso or Glu, are active when bound to a protein co-activator. The example we will discuss about this is the estrogen receptor.
The activation domain of the estrogen receptor has to be bound to estrogen to be in active conformation. The activation domain of the estrogen receptor is unstructured, then estrogen is bound in the pocket. The alpha helix of the receptor moves to interact with estrogen, and the other proteins move and fold, allowing the helix to contact another protein, a co-activator, which then leads to estrogen expression, also known as signalling.
Now, Tamoxifen is an antagonist or repressor in this process. When tamoxifen binds to the estrogen receptor, it blocks estrogen signals. The receptor does not undergo the necessarily structural change.
Another example is the model of the enhanceosome that forms on the B-interferon enhancer. Interferons are proteins made by host cells in response to the presence of pathogens such as viruses, bacteria, parasites, and tumour cells to signal the immune system to evoke a response.* Independent transcription factors bind together over a short sequence of the enhancer. The combination is called a multi-protein complex, which initiates transcription for a particular promotor. HMGI bends the minor groove of the DNA and then protein-protein interactions occur with highly cooperative binding to form a stable assembly of activators of transcription.
Transcription is activated through another complex known as a mediator complex, which forms a molecular bridge between the domains and Pol II to resolve the spatial separation between the activation region and promoter regions. This is a multi-protein conversation. The tail end interacts with the activation domain, and the head with Pol II. From there, transcription is initiated of the gene.
*For further details on the immune system, stay tuned! I will be covers a series of posts about the innate and adaptive immune responses including plenty of information about the roles of the various cells and their significance.
The activation domain of the estrogen receptor has to be bound to estrogen to be in active conformation. The activation domain of the estrogen receptor is unstructured, then estrogen is bound in the pocket. The alpha helix of the receptor moves to interact with estrogen, and the other proteins move and fold, allowing the helix to contact another protein, a co-activator, which then leads to estrogen expression, also known as signalling.
Now, Tamoxifen is an antagonist or repressor in this process. When tamoxifen binds to the estrogen receptor, it blocks estrogen signals. The receptor does not undergo the necessarily structural change.
Another example is the model of the enhanceosome that forms on the B-interferon enhancer. Interferons are proteins made by host cells in response to the presence of pathogens such as viruses, bacteria, parasites, and tumour cells to signal the immune system to evoke a response.* Independent transcription factors bind together over a short sequence of the enhancer. The combination is called a multi-protein complex, which initiates transcription for a particular promotor. HMGI bends the minor groove of the DNA and then protein-protein interactions occur with highly cooperative binding to form a stable assembly of activators of transcription.
Transcription is activated through another complex known as a mediator complex, which forms a molecular bridge between the domains and Pol II to resolve the spatial separation between the activation region and promoter regions. This is a multi-protein conversation. The tail end interacts with the activation domain, and the head with Pol II. From there, transcription is initiated of the gene.
*For further details on the immune system, stay tuned! I will be covers a series of posts about the innate and adaptive immune responses including plenty of information about the roles of the various cells and their significance.
DNA Binding Domains of Transcription Factors
Overview: Details about the various DNA binding domains of transcription factors including about their structure, function, and ways of ensuring sufficient regulatory diversity.
Transcription factors whether activator or repressor have DNA-binding domains with structural motifs that bind specific DNA sequences. The binding commonly results from noncovalent interactions, between the alpha helices of the protein's DNA binding domain and atoms on the bases in the major groove of the DNA helix. The positively charged amino acids can interact with the with the negatively charged backbone of the DNA. Hence, this is a charged-based, or ionic, interaction. Several different motifs of DNA-binding domains are known.
Zinc-Finger Motifs
In brief, these are protein regions that fold around a central Zn+2 ion.
C2H2 zinc finger:
These are most commonly found in humans. The binding of the zinc ion occurs by two cysteine and two histidine residues in the domain, which allow the insertion of the a-helix (a barrel-like structure) into the major groove of the DNA of the enhancer element. They contain 3 or more repeating finger units and bind as monomers to the DNA sequence. Since a monomer is only one protein, there is no symmetry.
C4 zinc finger:
These are only found in around 50 human transcription factors of the nuclear receptor family. Nuclear receptors are a class of proteins which are found in cells and sense steriod and thyroid hormones. Unlike C2H2 zinc fingers, C4 zinc fingers contain only two finger units, and bind as homodimers - two identical macromolecules. This allows them to have two-fold rotational symmetry, meaning the second is a mirror image of itself, and they bind to consensus DNA sequences that are inverted repeats.
Other Types of DNA-Binding Domains
Leucine-zipper:
They are characterized by having a leucine residue in every seventh position of the protein sequence, therefore they exhibit periodicity. Since, the protein has hydrophobic amino acid interactions, two proteins will interact in a coil-coil domain region, with leucine on either side to form a hydrophobic pocket, giving it the stability to dimerize. What is important to remember about leucine-zippers is that they are heterodimers (two different proteins) with basic amino acids possessing an enriched net positive charge that are capable of interacting with the phosphate backbone of the major groove of DNA. Also, Leucine-zippers belong to a larger family of related proteins known as "basic zippers" or bZip, which have similar hydrophobic interactions, but just with different hydrophobic amino acids.
Basic Helix-Loop-Helix (bHLH)
In structure, the Basic Helix-Loop-Helix is similar to the bZip, however a nonhelical loop separates two a-helical regions. This non-helical region doesn't not necessarily have a function, it is just a structural feature of which to be aware. Different bHL proteins can also form heterodimers.
A more interesting topic and question to consider is the following (drum roll please):
How can the finite set of transcription factors generate enough regulatory diversity?
Well, in a few ways. By the use of heterodimers, inhibitory factors, and cooperative binding. Please note that this list is probably not all inclusive, full meal combo done deal.
1) Heterodimers
Since each monomer has different DNA-binding specificity, combining two different monomers to make a heterodimer, increases diversity by becoming a problem of combinatorics, which is pretty sweet.
2) Inhibitory factors
The protein can block DNA binding by using some bZip and bHLH monomers, preventing expression of genes. Also, the inhibitory factor interacts with the transcription factor, preventing it from forming a homodimer or heterodimer with factors available.
3) Cooperative Binding
Two independent transcription factors with different protein complexes bind to their independent transcription elements, which are in close proximity to each other. Separately they have weak affinity to DNA, but when both are present, they share strong cooperative binding at the enhancer region.
Transcription factors whether activator or repressor have DNA-binding domains with structural motifs that bind specific DNA sequences. The binding commonly results from noncovalent interactions, between the alpha helices of the protein's DNA binding domain and atoms on the bases in the major groove of the DNA helix. The positively charged amino acids can interact with the with the negatively charged backbone of the DNA. Hence, this is a charged-based, or ionic, interaction. Several different motifs of DNA-binding domains are known.
Zinc-Finger Motifs
In brief, these are protein regions that fold around a central Zn+2 ion.
C2H2 zinc finger:
These are most commonly found in humans. The binding of the zinc ion occurs by two cysteine and two histidine residues in the domain, which allow the insertion of the a-helix (a barrel-like structure) into the major groove of the DNA of the enhancer element. They contain 3 or more repeating finger units and bind as monomers to the DNA sequence. Since a monomer is only one protein, there is no symmetry.
C4 zinc finger:
These are only found in around 50 human transcription factors of the nuclear receptor family. Nuclear receptors are a class of proteins which are found in cells and sense steriod and thyroid hormones. Unlike C2H2 zinc fingers, C4 zinc fingers contain only two finger units, and bind as homodimers - two identical macromolecules. This allows them to have two-fold rotational symmetry, meaning the second is a mirror image of itself, and they bind to consensus DNA sequences that are inverted repeats.
Other Types of DNA-Binding Domains
Leucine-zipper:
They are characterized by having a leucine residue in every seventh position of the protein sequence, therefore they exhibit periodicity. Since, the protein has hydrophobic amino acid interactions, two proteins will interact in a coil-coil domain region, with leucine on either side to form a hydrophobic pocket, giving it the stability to dimerize. What is important to remember about leucine-zippers is that they are heterodimers (two different proteins) with basic amino acids possessing an enriched net positive charge that are capable of interacting with the phosphate backbone of the major groove of DNA. Also, Leucine-zippers belong to a larger family of related proteins known as "basic zippers" or bZip, which have similar hydrophobic interactions, but just with different hydrophobic amino acids.
Basic Helix-Loop-Helix (bHLH)
In structure, the Basic Helix-Loop-Helix is similar to the bZip, however a nonhelical loop separates two a-helical regions. This non-helical region doesn't not necessarily have a function, it is just a structural feature of which to be aware. Different bHL proteins can also form heterodimers.
A more interesting topic and question to consider is the following (drum roll please):
How can the finite set of transcription factors generate enough regulatory diversity?
Well, in a few ways. By the use of heterodimers, inhibitory factors, and cooperative binding. Please note that this list is probably not all inclusive, full meal combo done deal.
1) Heterodimers
Since each monomer has different DNA-binding specificity, combining two different monomers to make a heterodimer, increases diversity by becoming a problem of combinatorics, which is pretty sweet.
2) Inhibitory factors
The protein can block DNA binding by using some bZip and bHLH monomers, preventing expression of genes. Also, the inhibitory factor interacts with the transcription factor, preventing it from forming a homodimer or heterodimer with factors available.
3) Cooperative Binding
Two independent transcription factors with different protein complexes bind to their independent transcription elements, which are in close proximity to each other. Separately they have weak affinity to DNA, but when both are present, they share strong cooperative binding at the enhancer region.
Friday, 5 October 2012
Locating Transcription Factors
Overview
This post is going to cover a bit about locating the transcription factors, purifying the protein, and then running some tests in vitro and in vivo to prove that yes, this protein really does aid in initiating transcription.
Particular sequences on DNA, known as transcriptional control elements, enhance transcription. They are called enhancers. Regulatory proteins called transcription factors bind to the enhancers, helping to initiate transcription of a gene. However, for this post we are not too concerned about the process of initiating transcription. Our goal is to identify and isolate these proteins (transcription factors) in the cell. How do we do this? There are three different methods that I know of (probably more exist) which shall be discussed. These techniques can be used together to purify the transcription factors desired.
1) Chromatography.
The basic idea behind the chromatography technique is that you pass a sample solvent of various proteins through a column of solid chromatography matrix and collect the protein in a test tube below after a certain time has elapsed. Different proteins will travel through the solid matrix at different rates depending on their properties. Here are three major types of chromatography used.
a) Ion-Exchange Chromatography
The solid matrix in this case is made up of positively charged beads. As the solvent passes through the matrix, the negatively-charged proteins will bind to the positively charged beads, and hence, will pass through the column more slowly. The negatively charged proteins will emerge first. To remove the positively charged proteins from the matrix, a salt is applied. The negative salt ions will bind to the positively charged matrix, displacing the negatively charged proteins.
* separation by charge
b) Gel-Filtration Chromatography
This time the solid matrix consists of porous beads which trap the smaller proteins, retarding them through the column. The large proteins pass through unhindered. The smaller proteins will eventually come out at the other end, a longer time will have just elapsed.
* separation by size
Step a) and b) can be done in interchangeable order, but step c) is always the last step.
c) Affinity Chromatography
This type of chromatography is the most specific type of chromatography. The beads are marked with a covalently attached sequence (or substrate in the case of an enzyme) specific to the protein which will bind and trap the desired protein molecule as it passes through the matrix. The rest of the proteins will pass through. To extract the desired protein, the nuclear extract is incubated with the beads, washed, and then eluted with increasing salt concentration. This method gives the highest purity of the protein.
2) DNase I Footprinting Experiments
These are useful as they reveal specific binding sites for DNA binding proteins and can be used as an assay in transcription factor purification. The DNA is end-labeled (as a point of reference) and then is partially digested with a nuclease like a restriction enzyme that cuts the DNA randomly. We take a strand of DNA without the DNA-binding protein and, the restriction enzyme will cut wherever it pleases creating numerous fragments. However, when we let the DNA-binding protein bind to its binding site on the DNA, then the restriction enzyme will cut everywhere except where the protein is bound. This allows you to know the protein's specific sequence. Now, if we pass this sample through an electrophoresis gel, the place where the protein bound will have no lines, because there will be no fragments at those base pairs.
3) EMSA (gel-shift, or band-shift assay)
The EMSA technique is better than footprinting for quantitative analysis of DNA-binding proteins, as to whether they bind or not to the DNA. However, it does not provide the specific DNA-binding sequence. When the DNA is passed through the gel, it is radiolabelled and contains a known regulatory element. The control is called a probe, and it will have a bound protein to its DNA fragment. The bound protein will cause the probe to migrate more slowly through the gel than DNA alone. To find whether the protein has bound to other strands of DNA, a band will show on the gel, whereas unbound DNA will pass through the gel quickly to the other side. This test is done with different DNA sequences from the same chromosome and the same proteins to find the binding sequence, although the specific base pairing cannot be isolated from this technique.
A Note about Transcription Factors
Transcription factors are proteins that stimulate or repress transcription by binding to promoter-proximal elements (a type of enhancer close to the promoter binding site, the TATA box), enhancers, and repressors in eukaryotic DNA. The modular protein structure of a transcription factor has a single DNA binding domain and one or more activation domains or repression domains, depending on whether the transcription factor is an activator or a repressor respectively.
Now that we have purified the protein, we need to prove that it does aid in initiating transcription indeed.
1) In Vitro Assay for Transcription Activity
In an example using SP1 as the transcription factor and Adenovirus DNA and SV40 DNA, Adenovirus is used as the control, as it does not require SP1 to initiate transcription. With or without SP1, transcription will occur. SV40, however, will not have any transcription if SP1 is not present. If SP1 is provided, transcription happens. Transcription is shown on these tests as a black band.
2) In Vivo Assay for Transcription Activity
This test is more powerful than the above in vitro assay because if something happens in a test tube doesn't mean it necessarily happens in the cell. Inserted into the cell, is a plasmid 1 containing the sequence required for the cell to transcribe the protein transcription factor being tested. Also, a plasmid 2 is inserted containing the binding sequence for the transcription factor along with reporter gene, which is just a gene which has an obvious effect upon the cell when transcribed. This is a co-transfection assay. If the transcription factor is truly a transcription factor, the reporter gene will be expressed in the cell. The assay can identify repressors as well as activators, and is a necessary method in cells that lack or do not express the gene encoding the protein to be tested.
A Note about Transcriptional Activators
Activators are modular proteins that have distinct functional domains including a DNA binding domain and an activation domain, which interacts with other proteins to stimulate transcription.
Activators were demonstrated in an experiment where two plasmids were again inserted into a cell. A variety of lengths of sequences for the gene that contained information to encode the transcription factor GAL4 in plasmid 1 were taken. Then it was measured whether or not the LacZ reporter gene was transcribed from the plasmid 2.
This example is a little bit trippy, because we are measuring the effects of transcription by how the cell transcribed the inserted plasmid 1 sequence of the transcription factor by (wait for it), measuring the transcription factor's initiation of transcription of the reporter gene in plasmid 2. Phewf!
The results examined were the binding to UASgal enhancer site on plasmid 2, and the B-galactosidase activity in the cell. When neither was shown in the deletion of base pair for the sequence of GAL4, the conclusion was that that was a DNA binding domain. No DNA binding domain present meant no binding to the enhancer site was possible, hence no transcription. However, with subsequent deletion of bases in plasmid 1, binding to UASgal was found, but little to no B-galactosidase activity, demonstrating the activation domains. This was significant because it revealed the activation domains to be in separate locations on the coding sequence than the DNA binding domain.
Activation domains can be in front of or behind the DNA binding domain. There can be multiple activation domains, but there is always only one DNA binding domain. If the domains are swapped around or fused together, a functional protein still results. They don't seem to be too fussy.
A Note About Transcriptional Repressors
As you might guess, they are the functional converse of activators. Most are modular proteins with a DNA-binding domain and a repression domain, which function by interacting with other proteins.
This post is going to cover a bit about locating the transcription factors, purifying the protein, and then running some tests in vitro and in vivo to prove that yes, this protein really does aid in initiating transcription.
Particular sequences on DNA, known as transcriptional control elements, enhance transcription. They are called enhancers. Regulatory proteins called transcription factors bind to the enhancers, helping to initiate transcription of a gene. However, for this post we are not too concerned about the process of initiating transcription. Our goal is to identify and isolate these proteins (transcription factors) in the cell. How do we do this? There are three different methods that I know of (probably more exist) which shall be discussed. These techniques can be used together to purify the transcription factors desired.
1) Chromatography.
The basic idea behind the chromatography technique is that you pass a sample solvent of various proteins through a column of solid chromatography matrix and collect the protein in a test tube below after a certain time has elapsed. Different proteins will travel through the solid matrix at different rates depending on their properties. Here are three major types of chromatography used.
a) Ion-Exchange Chromatography
The solid matrix in this case is made up of positively charged beads. As the solvent passes through the matrix, the negatively-charged proteins will bind to the positively charged beads, and hence, will pass through the column more slowly. The negatively charged proteins will emerge first. To remove the positively charged proteins from the matrix, a salt is applied. The negative salt ions will bind to the positively charged matrix, displacing the negatively charged proteins.
* separation by charge
b) Gel-Filtration Chromatography
This time the solid matrix consists of porous beads which trap the smaller proteins, retarding them through the column. The large proteins pass through unhindered. The smaller proteins will eventually come out at the other end, a longer time will have just elapsed.
* separation by size
Step a) and b) can be done in interchangeable order, but step c) is always the last step.
c) Affinity Chromatography
This type of chromatography is the most specific type of chromatography. The beads are marked with a covalently attached sequence (or substrate in the case of an enzyme) specific to the protein which will bind and trap the desired protein molecule as it passes through the matrix. The rest of the proteins will pass through. To extract the desired protein, the nuclear extract is incubated with the beads, washed, and then eluted with increasing salt concentration. This method gives the highest purity of the protein.
2) DNase I Footprinting Experiments
These are useful as they reveal specific binding sites for DNA binding proteins and can be used as an assay in transcription factor purification. The DNA is end-labeled (as a point of reference) and then is partially digested with a nuclease like a restriction enzyme that cuts the DNA randomly. We take a strand of DNA without the DNA-binding protein and, the restriction enzyme will cut wherever it pleases creating numerous fragments. However, when we let the DNA-binding protein bind to its binding site on the DNA, then the restriction enzyme will cut everywhere except where the protein is bound. This allows you to know the protein's specific sequence. Now, if we pass this sample through an electrophoresis gel, the place where the protein bound will have no lines, because there will be no fragments at those base pairs.
3) EMSA (gel-shift, or band-shift assay)
The EMSA technique is better than footprinting for quantitative analysis of DNA-binding proteins, as to whether they bind or not to the DNA. However, it does not provide the specific DNA-binding sequence. When the DNA is passed through the gel, it is radiolabelled and contains a known regulatory element. The control is called a probe, and it will have a bound protein to its DNA fragment. The bound protein will cause the probe to migrate more slowly through the gel than DNA alone. To find whether the protein has bound to other strands of DNA, a band will show on the gel, whereas unbound DNA will pass through the gel quickly to the other side. This test is done with different DNA sequences from the same chromosome and the same proteins to find the binding sequence, although the specific base pairing cannot be isolated from this technique.
A Note about Transcription Factors
Transcription factors are proteins that stimulate or repress transcription by binding to promoter-proximal elements (a type of enhancer close to the promoter binding site, the TATA box), enhancers, and repressors in eukaryotic DNA. The modular protein structure of a transcription factor has a single DNA binding domain and one or more activation domains or repression domains, depending on whether the transcription factor is an activator or a repressor respectively.
Now that we have purified the protein, we need to prove that it does aid in initiating transcription indeed.
1) In Vitro Assay for Transcription Activity
In an example using SP1 as the transcription factor and Adenovirus DNA and SV40 DNA, Adenovirus is used as the control, as it does not require SP1 to initiate transcription. With or without SP1, transcription will occur. SV40, however, will not have any transcription if SP1 is not present. If SP1 is provided, transcription happens. Transcription is shown on these tests as a black band.
2) In Vivo Assay for Transcription Activity
This test is more powerful than the above in vitro assay because if something happens in a test tube doesn't mean it necessarily happens in the cell. Inserted into the cell, is a plasmid 1 containing the sequence required for the cell to transcribe the protein transcription factor being tested. Also, a plasmid 2 is inserted containing the binding sequence for the transcription factor along with reporter gene, which is just a gene which has an obvious effect upon the cell when transcribed. This is a co-transfection assay. If the transcription factor is truly a transcription factor, the reporter gene will be expressed in the cell. The assay can identify repressors as well as activators, and is a necessary method in cells that lack or do not express the gene encoding the protein to be tested.
A Note about Transcriptional Activators
Activators are modular proteins that have distinct functional domains including a DNA binding domain and an activation domain, which interacts with other proteins to stimulate transcription.
Activators were demonstrated in an experiment where two plasmids were again inserted into a cell. A variety of lengths of sequences for the gene that contained information to encode the transcription factor GAL4 in plasmid 1 were taken. Then it was measured whether or not the LacZ reporter gene was transcribed from the plasmid 2.
This example is a little bit trippy, because we are measuring the effects of transcription by how the cell transcribed the inserted plasmid 1 sequence of the transcription factor by (wait for it), measuring the transcription factor's initiation of transcription of the reporter gene in plasmid 2. Phewf!
The results examined were the binding to UASgal enhancer site on plasmid 2, and the B-galactosidase activity in the cell. When neither was shown in the deletion of base pair for the sequence of GAL4, the conclusion was that that was a DNA binding domain. No DNA binding domain present meant no binding to the enhancer site was possible, hence no transcription. However, with subsequent deletion of bases in plasmid 1, binding to UASgal was found, but little to no B-galactosidase activity, demonstrating the activation domains. This was significant because it revealed the activation domains to be in separate locations on the coding sequence than the DNA binding domain.
Activation domains can be in front of or behind the DNA binding domain. There can be multiple activation domains, but there is always only one DNA binding domain. If the domains are swapped around or fused together, a functional protein still results. They don't seem to be too fussy.
A Note About Transcriptional Repressors
As you might guess, they are the functional converse of activators. Most are modular proteins with a DNA-binding domain and a repression domain, which function by interacting with other proteins.
Tuesday, 2 October 2012
Thanksgiving Specials
Does Bagel Trance and Happenstance have Thanksgiving specials you asked?
Oh but of course!
Intended upcoming posts (perhaps not in this order) include:
1) The immune system
2) Some details about transcription and genomics (three posts, on three topics)
3) Finding and isolating the transcription factor
4) (Very) General information about blood
5) Bodily Fluids
6) Transport Mechanisms
7) Taking Ancient Greek and other introductory classical language courses
8) The history of Racemic Mixtures and polarimetry
9) Ideal Gases in thermodynamics
Of course, this be more material than Thanksgiving weekend can handle! :)
** I would love to do a series of posts about organic chem, however most of my studying in this course has been very "try the problems and build the models and discuss/argue with friends while trying the problems and building the models." This approach is a bit hard to capture by writing, as it is very hands-on.
Oh but of course!
Intended upcoming posts (perhaps not in this order) include:
1) The immune system
2) Some details about transcription and genomics (three posts, on three topics)
3) Finding and isolating the transcription factor
4) (Very) General information about blood
5) Bodily Fluids
6) Transport Mechanisms
7) Taking Ancient Greek and other introductory classical language courses
8) The history of Racemic Mixtures and polarimetry
9) Ideal Gases in thermodynamics
Of course, this be more material than Thanksgiving weekend can handle! :)
** I would love to do a series of posts about organic chem, however most of my studying in this course has been very "try the problems and build the models and discuss/argue with friends while trying the problems and building the models." This approach is a bit hard to capture by writing, as it is very hands-on.
Physical Chemistry Mock Midterm Answer Key
Disclaimer: Use at your own risk. The definitions, short answers, and calculations may not be correct or fully correct. Hope it is helpful and please leave comments if you have found any discrepancies. Also, while doing this exam, I discovered many questions were poorly phrased for which I apologize. (One mistake has already been caught by somebody! Please help me catch more!)



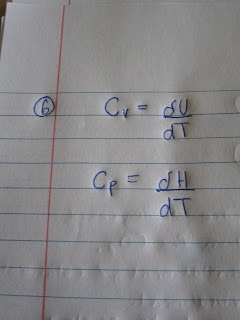
Physical Chemistry Mock Midterm Exam
**disclaimer: I have
no idea exactly what will be on the midterm exam beyond what was listed by the professor in class, although I
do know these section headings are accurate. This mock midterm is written only as
a study tool. It’s usefulness is debatable.
Part I:
Definitions (20%)
Provide definitions for each of the following terms. Do NOT
give any examples of the terms or you will lose marks. (1 mark per definition, -1 mark if
example given)
1) System: the part of the universe of interest
2) Isolated system: system that exchanges neither matter nor
energy with the surrounding
3) closed system: system that exchanges matter but not
energy with the surroundings
4) open system: system that exchanges both matter and energy
with the surroundings
5) surroundings: the rest of the universe, excluding the
system or part of the universe of interest
6) Zeroth Law of Thermodynamics: If A is in thermal
equilibrium with B and B is in thermal equilibrium with C, then A is in thermal
equilibrium with C
7) Thermal Energy: the energy around us in the
translational, rotational, vibrational states
8) Internal Energy: the energy needed to create the system
from atoms, the thermal energy (kinetic energy of motion – moving, rotational
and vibration) and bond energy – the electrostatic energy of the attraction and
repulsion of nuclei, does not include PV work
9) Heat: transfer of energy by conduction, radiation,
convenction; path dependent
10) Work: energy transferred by a force acting on a
distance; path dependent
11) First Law of Thermodynamics: delta U = q + w, where q is
the heat added to the system, and w is the work done on the system, and delta U
is the change in internal energy
12) Reversible Path: a path that depends only on the initial
and final states of the system
13) Nonreversible Path: a path that depends on the method
used to achieve the final state from the initial state
14) State Function: a property whose change is only
dependent on the initial and final states of a system
15) Heat Capacity (also include definition for molar heat
capacity and specific heat capacity): the amount of heat required to raise the temperature by 1K,
dependent on the translational, rotational and vibrational states of the
molecules
molar heat capacity: the amount of heat required to raise
the temperature by 1K per 1 mol of substance
specific heat capacity: the amount of heat required to raise
the temperature by 1K per 1 kg of substance
16) Translational States: this in my humble opinion, is an unfair
question as the mathematics involving this definition go back to particle in a
box and are a bit complicated.
A better question would be: How do you eliminate the
translational states of a molecule?
Answer: By taking a frame of reference.
17) Rotational States: same as angular momentum, denoted by
Jmj where J goes from 0 to infinity, -J < mj < +J
18) Vibrational States: given in integer values from 0 to
infinity
19) Equiparition of Energy: the division of energy amongst
all of the degrees of freedom of the molecule (this definition may be
incomplete, however with the content of the course thus far, and its not
rigorous nature, I suspect it may be ok)
20) Bond Energy: the energy required to break a chemical
bond into its constituents
21) Hess’s Law: state functions do not depend on the path
22) Heat of Formation of a Molecule: the heat required to
form a molecule from its elements
22) Heat of Combustion of a Molecule: the heat required to
burn a molecule (react with gaseous oxygen) to usually produce gaseous carbon
dioxide and liquid water
23) Enthalpy: H = U + PV where U is the internal energy of a
system, P is the pressure of the surroundings, and v is the volume of a system
26) State: the parameters that are measured and describe a
system under those conditions
Part II:
Calculations
Provide all work, including appropriate equations, notation,
and manipulation.
Try (or retry) calculations from the textbook without
looking at the solutions manual.
For a good selection of questions do:
Energy, Heat, Work
3, 4, 6, 10 (this question might require you to do 9 or something, sorry)
Thermochemistry
15, 17, 21, 24, 29, 32, 34
Equiparitioning of
Energy
1)
Calculate the number of degrees of freedom for
carbon dioxide. Estimate the molar heat capacity of the substance.
2)
Calculate the number of degrees of freedom for
methane. Estimate the molar heat capacity of the substance.

Part III: Short
Answers
1)
What type of system is a coffee cup calorimeter?
What type of system is a bomb calorimeter? Compare and contrast.
A coffee cup calorimeter is a closed system. Matter cannot
leave the system, but energy can. A bomb calorimeter is an isolated system, so
both matter and energy cannot leave the system.
2)
If system A is 25ºC and system B is 25ºC, and
system C is in equilibrium with system B, what can we conclude about the
temperature of system C and its relationship with system A?
Using the Zeroth Law of Thermodynamics, we know that since system C is in equilibrium with system B, it must have the same temperature of 25ºC. System C may be in thermal equilibrium with system A if it is in contact with system A, or if it is in contact with system B and system B is in contact with system A. But the problem doesn't include enough information to make a solid conclusion about the relationship of system C and A.
3)
Draw potential energy diagram of a diatomic
molecule. Label the place of greatest repulsion, A. Label the place of greatest
stability, B. Label zero point, C. Label and show the bond energy. Draw in the
largest state (rotational/vibrational/translational) , and discuss the
placement and magnitude of the other states
(rotational/vibrational/translational).

4)
Is the maximum work done by the system
reversible or irreversible? Explain referring to a P versus V diagram.
The maximum work done by the system is reversible. It is
done along the same isotherm (at constant temperature), and depends on only the
final and initial states, not the path. In an isothermal expansion or compression, the process is very slow, and so the work done by or on the system is replaced by the heat into or out of the system for a change of internal energy of zero. (There is a nice diagram on page 58 of the text that you could refer to with this answer.)
5)
Using the definition of enthalpy, show that it
is equal to the heat at constant pressure.

6)
Define both the heat capacity at constant volume
and the heat capacity at constant pressure.

7)
Is work a state function? Explain.
No, work is not a state function, as it is path dependent.
(Using a car, crane or helicopter to move an object all takes different amounts
of energy to reach the final state)
8)
Discuss how differential calorimetry finds the
unknown heat capacity of a substance.
Differential calorimetry compares a sample of unknown heat capacity with a sample of known heat capacity called the reference sample. This reference must be well-defined over a large range of temperatures, and not undergo a phase transition. Then you test the two samples. If the unknown undergoes a phase transition, a peak on a heat versus temperature graph will appear. Comparing this value with the value of the known heat capacity of the reference sample will aid in calculating the heat capacity of the unknown. qs - qr
9)
What is the standard enthalpy of formation of
oxygen gas?
As oxygen gas is in elemental form, its standard enthalpy of
formation is zero.
10)
Why
is the bond energy always positive?
The bond energy is the amount of energy required to break a
molecule into its constituents. Energy is usually required to put into the
system (positive) to break a chemical bond.
11)
Hydrogen gas only has 2 rotational degrees of freedom. Explain
why.
Hydrogen gas can rotate along the three axes. However, the x
and y axes exhibit much greater rotation for a linear molecule as does the z
axes, since the x and y axes contain the two atoms whereas the z axis does not
and so is not as greatly effected.
12)
Discuss the effects of temperature when comparing calculated
heat capacities using equiparitioning of energy and the actual measured heat
capacity values.
At warmer temperatures, there is greater agreement of the
calculated heat capacities with the actual heat capacity values. At colder
temperatures there is less agreement between the calculated and actual heat
capacities. This is because the vibrational molecules are more widely spaced
and energy cannot move as freely between vibrational modes, so a transition requires
a more significant (or sufficiently large quantum) amount of energy.
Part IV: True or
False
This section will NOT
be on the exam, however, it may provide a good snapshot as to whether the
definitions are understood.
1)
Heat leaves the system. q is positive. True/False q is negative.
2)
Energy is stored in chemical bonds. True/false
energy is stored in the degrees of freedom of a molecule
3)
The system does work. w is negative. True/false
4)
Internal energy does not depend on PV work. True/false
5)
Bond energy is usually greater than thermal
energy. True/false
6)
A diatomic gas has 2 degrees of translational
energy. True/false
A diatomic gas has 3 degrees of translational energy and 2 degrees of
rotational energy.
7)
PV work is positive for an expansion of a gas. True/false
PV work is negative for the expansion of a gas and positive for the
compression of a gas.
8)
The lowest vibrational state is at the zero
point. True/false The lowest vibrational state is
slightly above the zero point.
9)
The heat capacity at a phase change is infinite. True/false
10) Vibrational states are the same as angular momentum. True/false
Rotational states are the same as angular momentum.
Part V: Bonus
Questions
1)
Show that PV=nRT is a state function using
partial derivatives and Euler’s Theorem for Exactness without referring to
Appendix C. (Just kidding! Bad joke I know.)
Refer to post titled “Friday Night State Functions.” But seriously we don't have to know this.
2)
Describe your experience and opinion of the
textbook with regards to whether you have the e-book or hardcopy, its
acquisition and availability, its use as a study tool, and to what level it
corresponds to and complements the lectures.
I have heard many (at least 5 opinions not my own) nuanced
responses to this question, and they all have strikingly similar themes.
Subscribe to:
Posts (Atom)